New position statement published by the IVD expert panel on influenza viruses
MDx CRO, an IVD consultancy and CRO company, reviews the latest position paper published by the EU Commission IVD Expert Panel. The publication discusses the risk classification of influenza assays under the In Vitro Diagnostic Regulation (IVDR) and focuses on whether influenza viruses should be considered as high-risk Class D pathogens or fall under other risk categories.
Background
Influenza viruses, like many respiratory viruses, have the potential to cause life-threatening diseases with a high or suspected high risk of propagation. The severity and transmissibility of an influenza virus strain depend on various factors, including virus-specific, host-specific, and environmental factors. Seasonal influenza strains undergo antigenic drift, while antigenic shift can result in new influenza A subtypes, potentially leading to pandemics.
Transmissibility and Severity
Transmissibility is a key indicator of the ease of movement of the influenza virus between individuals and communities. It is influenced by the virus’s ability to spread from person to person, transmission dynamics, and population susceptibility. Influenza viruses can be transmitted not only between humans but also from animals to humans and vice versa, posing public health risks.
Disease Severity of Influenza A(H1N1)pdm09
The A(H1N1)pdm09 strain, responsible for the 2009 pandemic, was remarkably different from seasonal influenza strains, leading to a higher fatality rate and impacting younger populations more severely. Since the pandemic, A(H1N1)pdm09 has continued to circulate, causing significant disease burden globally. Despite available vaccines, their effectiveness remains a concern, and oseltamivir-resistant variants have been reported. Ongoing global surveillance and analysis of antiviral susceptibility are crucial for public health and patient care.
Other Circulating Influenza Virus Strains
Various seasonal influenza viruses, both type A and B, have the potential for high transmissibility and severe disease. An unusual and severe epidemic was observed during the 2017-2018 season, primarily dominated by influenza B virus, while influenza A(H3N2) and A(H1N1)pdm09 also caused severe cases in older adults. Additionally, zoonotic transmission of novel influenza strains, like H3N2, H7N9, and H5N6, has been reported, highlighting the importance of global surveillance and early warning systems.
Feasibility of Developing General Common Specifications for influenza assays
Given the unpredictable changes in influenza strains and the potential for novel variants to emerge, it is challenging to develop general common specifications with minimum performance requirements for class D devices. Assays for seasonal influenza detection, intended for individual infection detection, may not be suitable for detecting emerging strains with pandemic potential or from non-human origins. Such assays may require different safety protocols and risk assessments, and rapid confirmatory real-life performance evaluation studies should be considered.
Risk classification of influenza assays under the IVDR
Influenza viruses, particularly A(H1N1)pdm09, have demonstrated the potential for high transmissibility and severe disease, leading to significant public health consequences. The development of general common specifications for all influenza strains is challenging due to the varying risks and objectives associated with different assay types. With regards to the risk classification of influenza assays under the IVDR, the Commission’s position paper points out that seasonal influenza assays should be classified as class C devices, while class D devices may be necessary for assays intended to detect strains with pandemic potential or from non-human origins. This position statement may contradict some of industry’s initial assumptions on the classification of devices for seasonal influenza. V2 of the MDCG guidance on classification rules for IVDs originally stated that a “device intended for the detection of influenza A/B virus (non pandemic)” was a class B device according to rule 6.
Implications for manufacturers of influenza assays
The classification of IVDs can be highly intricate, and determining whether a device should be classified as Class B or Class C (and even D) often presents challenges. This complexity is particularly evident in devices where the associated risk level varies between low, moderate, and high, depending on factors such as circulating strains and the intended use of the device.
Notified Bodies have been adhering to the MDCG classification guidance to classify these devices and grant appropriate certification according to the risk class. For instance, assays designed to detect seasonal influenza were typically considered Class B devices, following examples provided in the guidance. On the other hand, assays intended for detecting high-risk strains would be classified as Class D (also according to the guidance).
This classification approach seemed logical until the latest advice from the expert panel was published. According to this updated advice, seasonal influenza assays could potentially be reclassified as Class C devices. This reclassification would have significant implications for manufacturers, as it would require the creation of Periodic Safety Update Reports (PSURs), Summary of Safety and Performance (SSP) documents, and other additional scrutiny throughout the entire process.
Given these potential changes, MDx CRO and relevant stakeholders are intrigued to learn how Notified Bodies will respond to this new development and what their expectations will be moving forward. Manufacturers may need to adapt their strategies to comply with a potential revised classification and the increased requirements, which may present both challenges and opportunities in the IVD industry.
Ongoing global surveillance, international collaboration, and data sharing are essential for effective influenza control and preparedness.
MDx CRO has extensive experience in conducting clinical performance studies and providing regulatory support for high-risk infectious disease IVDs.
Senior regulatory leader and former BSI IVDR reviewer with deep experience in CE marking high-risk IVDs, companion diagnostics, and IVDR implementation.
IVD Consultancy: Balancing Expertise & Cost-Efficiency with MDx CRO
July 27, 2023
Striking a balance between expertise and cost-efficiency in IVD consultancy can be a tricky task, especially within the European Union’s context, where there’s a notable dearth of specialized consultants. Recognizing this challenge and its impact on your business’s success is what we do at MDx CRO. This article offers a deep dive into the crucial factors to bear in mind when picking your IVD consultant and how MDx CRO combines expertise and cost-effectiveness smoothly.
Selecting a consultant should be a process that considers a variety of factors aligned with your company’s unique needs and budget. Here’s a look at some of the most important aspects, including domain expertise, relevance to the industry, cost-effectiveness, and cultural fit.
Depth of Expertise
At the heart of a successful IVD consultancy engagement is a consultant’s profound and extensive knowledge. This is especially important in a specialized area like IVD, where the regulatory terrain is complex and quite different from general medical devices. Scrutinize potential consultants’ qualifications, industry experience, and roles within IVD companies, regulatory bodies, and regulators to ascertain they possess the necessary domain expertise. At MDx CRO, we value expertise and consider its cost justified by the immense value it brings to your project. Our consultants are not just well-versed in the IVD sector; they are seasoned professionals with a wealth of practical experience. A common error is opting for a consultancy firm with a general understanding, perhaps in areas like pharma or general medical devices. If you’re not engaging with a partner and consultants with a deep understanding of your specific IVD technology, you’re essentially laying down hurdles in your own path.
Industry Relevance and Recommendations
It’s crucial to check how up-to-date and applicable the consultant’s knowledge is. Review their published works, evaluate their communication style, and verify if their insights align with your needs. Moreover, consider seeking industry colleagues’ recommendations or checking professional networks like LinkedIn. A reputable consultant should be able to give a rundown of their services, providing a clear view of their value proposition and balance of cost and benefits.
Comprehensive Service Offering and Cost-Efficiency
Depending on your market entry strategy, you might require a consultant with expertise in your target regions. Consultants adept in navigating both EU and US regulatory landscapes can deliver significant value, providing cost-efficiency by producing uniform documentation for multiple markets. This saves time and eliminates redundant tasks. At MDx CRO, we take pride in offering such comprehensive and cost-effective services, positioning ourselves as your all-in-one solution for IVD regulatory needs.
Cultural Compatibility
While often overlooked, a strong cultural fit plays a crucial role in a successful collaboration. An open conversation with potential consultants can help you gauge this compatibility in terms of language, time zone, and communication style. A good cultural fit promotes better collaboration, yielding more bang for your buck.
In conclusion, choosing the right IVD consultancy involves a careful balance of expertise, industry relevance, cost-effectiveness, and cultural fit. At MDx CRO, we embody this balance, providing comprehensive, cost-effective IVD consultancy services without compromising on value. Our goal is not only to deliver expert solutions but to do so at fair and transparent prices, distinguishing us in the IVD industry.
Feel free to contact us today to discuss your IVD consultancy needs. It’s where our IVD Consultancy Expertise and Cost-Efficiency come into play, ensuring that you receive the best possible value for your investment.
Written by:
Carlos Galamba
CEO
Carlos spent several years of his career at BSI, where he developed a global network of relationships with top EU Notified Bodies. As the first in-house clinician for IVDs at BSI, he led the implementation of the BSI clinical oversight process, providing hundreds of CE marking recommendations for IVDs and supporting IVDR Notified Body designations.
The recent amendment of the Medical Devices Regulation (MDR) and In Vitro Diagnostic Medical Devices Regulation (IVDR) through Regulation (EU) 2023/607 has introduced crucial changes to safeguard public health, prioritize patient safety, and prevent disruptions in healthcare services. The Q&A document regarding the extension of the MDR transition period and removal of the “sell-off” periods has been updated (July 2023) in response.
Key Changes to the Q&A Document on the MDR Transition Period
MDx CRO had previously published a comprehensive summary of the European Commission’s Q&A document.
The following questions in the Q&A document have undergone significant changes:
Q&A no 1: The Commission will provide flowcharts to assist manufacturers and other relevant parties in determining whether a device falls under the extended transitional period specified in Article 120 MDR.
Q&A no 2: Clarifies that a letter from a notified body regarding the certificate’s expiry or a controlled phase-out of production, mutually agreed upon by the notified body and the manufacturer before 20 March 2023, is not considered a certificate withdrawal.
Q&A no 7: Manufacturers need to provide a self-declaration confirming their compliance with the extension conditions and stating the end date of the transition period. They can include a “confirmation letter” from the notified body, which identifies the devices and certificates covered. Templates for the self-declaration and notified body’s confirmation letter are available at this link. Furthermore, an updated factsheet for competent authorities in non-EU/EEA countries explains the functioning of the extended transition period.
Q&A no 8: Clarifies that that when submitting information, notified bodies (as per Article 36(2) MDR) must have the capability to include the relevant (digital) document(s) in their own records. Simply having ‘read-only’ access to the manufacturer’s electronic data platform is not considered sufficient.
Q&A no 17: Manufacturers must inform the notified body about devices that require surveillance, especially if surveillance activities were discontinued due to certificate expiration before 20 March 2023. This information allows the notified body to conduct proper surveillance and make necessary arrangements with the manufacturer.
New Additions to the Q&A Document
The following questions have been newly introduced in the MDR Transition Period Q&A document:
Q&A no 6.1: If a competent authority grants a national derogation under Article 59 MDR or requires a manufacturer to follow the applicable conformity assessment procedure as per Article 97 MDR after 20 March 2023, the extended transitional period specified in Article 120(3a) MDR does not apply.
Q&A no 6.2: If the removal of the CE marking is a condition or consequence of the derogation granted by the competent national authority according to Article 59 of the MDR, the device may still be placed on the market with a CE marking, provided that all other conditions are met.
Q&A no 9.1: If a manufacturer withdraws the conformity assessment application or terminates the written agreement with the notified body after the deadlines, the extended transitional period ends. However, if the manufacturer switches to another notified body and fulfills all conditions, the transitional period continues. Updated documentation is necessary after the change, except when changing notified bodies due to non-compliance.
Q&A no 9.2: The manufacturer’s organization may undergo administrative changes, such as changes in name, address, or legal form, which generally do not impact the transitional period during the extended transition period. However, the transfer of devices from a manufacturer certified under MDD/AIMDD to another manufacturer intending to market them under MDR is not covered by the transitional period, unless both manufacturers are part of the same larger organization.
Q&A no 11.1: Legacy devices are not required to comply with the Unique Device Identification (UDI) requirements of the MDR during the extended transitional period. Even after May 26, 2024, when the manufacturer of the legacy device must have an MDR-compliant Quality Management System (QMS), UDI requirements will only apply if UDI assignment is necessary for those devices according to Article 10(9), point (h), of the MDR.
MDR Transition with MDx CRO
In conclusion, these recent changes and additions to the medical device regulations are significant milestones that harmonize industry standards and ensure a smooth transition for legacy devices, prioritizing safety and public health. The Q&A document serves as an essential tool for manufacturers, notified bodies, and competent authorities navigating the evolving regulatory landscape within the European Union.
Take advantage of MDx CRO’s expertise to ensure MDR compliance and meet transitional period requirements. Partnering with MDx CRO empowers manufacturers to meet safety standards, unlock opportunities in the European healthcare market, and contribute to healthcare advancements.
IVDR Compliance: Progress in EU Reference Laboratories and consequences for High-Risk IVDs
June 22, 2023
EU Commission evaluates EURLs
The European Commission is making significant progress in evaluating applications for EU reference laboratories (EURLs) to ensure compliance with the In Vitro Diagnostic Medical Devices Regulation (IVDR). These designated laboratories play a vital role in upholding the safety and efficacy of high-risk in vitro diagnostic medical devices (IVDs) within the European Union (EU).
In accordance with Article 100 of Regulation (EU) 2017/746, the Commission has been diligently assessing the applications received for EURL designation. The EURLs are entrusted with key responsibilities related to IVDR compliance, including advisory roles and activities associated with market access, specifically for class D devices that carry the highest level of risk. These laboratories are responsible for verifying the performance and ensuring the conformity of class D devices with common specifications, as well as conducting batch testing.
While no EURLs have been designated under Regulation (EU) 2017/746 thus far, the European Commission launched a call for applications in July 2022, targeting eight categories of class D devices. These categories encompass various areas such as hepatitis and retroviruses, herpesviruses, bacterial agents, arboviruses, respiratory viruses causing life-threatening diseases, haemorrhagic fever and other biosafety level 4 viruses, parasites, and blood grouping.
Candidate laboratories were invited to submit their applications to the national contact points in their respective Member States by January 2023. Subsequently, Member States were responsible for submitting the applications on behalf of the candidate laboratories to the European Commission, with a deadline of March 31, 2023.
8 Laboratories apply for designation
As of the deadline, the European Commission has received eight applications for review. These applications are currently undergoing a comprehensive assessment based on the specific criteria outlined in the call for applications. The evaluation process ensures that applicant laboratories meet the necessary standards and possess the combined capacity to handle the expected volume of requests related to market access tasks.
The European Commission aims to conclude its assessment process by the third quarter of 2023. However, it has been determined that none of the applicant laboratories in the category of haemorrhagic fever and other biosafety level 4 viruses possess the required combined capacity to adequately address the anticipated volume of requests. Consequently, no EU reference laboratory will be designated for this particular category following the current call for applications.
Despite the absence of a designated EURL, manufacturers can still pursue conformity assessment of their IVDs through notified bodies, allowing for certification and lawful placement of products on the EU market in adherence to Regulation (EU) 2017/746.
It is important to note that additional calls for EURLs in the field of IVDs may be issued in the future, and relevant information will be published in advance to facilitate preparedness among stakeholders.
For detailed guidance on the integration of EURLs into the conformity assessment process once they are designated, manufacturers are encouraged to refer to MDCG 2021-4.
The establishment of EU reference laboratories marks a significant stride towards enhancing IVDR compliance and ensuring the quality and safety of high-risk IVDs across the European Union. The European Commission’s thorough evaluation process and steadfast commitment to regulatory compliance are instrumental in safeguarding public health and fostering innovation in the field of in vitro diagnostics.
MDx CRO: Your Partner for High-Risk IVD Studies
Looking to navigate the stringent requirements of EU common specifications and EURL standards?
Partner with MDx CRO. Our expert team specializes in designing and conducting IVD studies that meet these rigorous demands.
We offer:
Customized Study Design: Tailored protocols aligned with high-risk IVD requirements, common specifications and EURL expectations.
ISO 20916 Clinical Performance studies in state-of-the-art EU laboratories: Accurate and reliable studies to demonstrate IVD clinical performance.
Regulatory Compliance Support: Expert guidance throughout the regulatory journey
Quality and Timeliness: Efficient execution, precise data collection, and timely reports.
With MDx CRO as your ally, achieve IVDR compliance and bring your high-risk IVDs to market confidently.
Contact us today to discuss your needs and ensure a predictable go-to-market strategy.
FAQs
Q: What tasks do EU Reference Laboratories (EURLs) perform?
A: EURLs have a range of important tasks designed to ensure effective IVDR compliance. These tasks include:
Verification of performance: EURLs verify the performance claimed by manufacturers and ensure compliance with Common Specifications or other solutions.
Testing of devices: EURLs perform tests on samples of manufactured class D devices or batches of class D devices to ensure their quality and safety.
Scientific and technical assistance: EURLs provide valuable scientific and technical assistance, opinions, and advice to support regulatory decision-making.
Network management: EURLs establish and manage networks and sub-networks of reference laboratories to facilitate collaboration and exchange of knowledge and expertise.
Development of testing methods: EURLs contribute to the development of appropriate testing and analysis methods for IVDs, promoting standardized practices.
Collaboration with notified bodies: EURLs collaborate with notified bodies to develop best practices and ensure consistency in conformity assessment procedures.
Recommendations on reference materials: EURLs provide recommendations on suitable reference materials and measurement procedures to enhance accuracy and reliability.
Contribution to standards development: EURLs actively participate in the development of Common Specifications (CS) and international standards to align regulatory requirements.
Q: Are EURLs only for class D devices?
A: While EURLs are primarily intended for class D devices, there is provision for an EU reference laboratory to be assigned for class C devices upon request from a Member State.
Q: What is the purpose of creating a network of laboratories across the European Union?
A: The aim is to establish a broad network of laboratories throughout the European Union to enhance the safety and compliance of the IVD market. These laboratories will adopt harmonized methods, ensuring coordinated processes, consistent testing protocols, and standardized reporting. They will also cooperate in quality assessment tests, develop joint guidelines, and coordinate the introduction of testing methods for emerging technologies.
Q: How will EURLs contribute to making the IVD market safer and compliant?
A: EURLs play a crucial role in verifying device performance, conducting rigorous testing, and providing scientific expertise. By adopting harmonized methods and collaborating within the network, EURLs ensure consistency, accuracy, and reliability in the assessment of IVDs. This ultimately contributes to a safer and more strictly IVDR compliance framework within the European Union.
Written by:
Carlos Galamba
CEO
Senior regulatory leader and former BSI IVDR reviewer with deep experience in CE marking high-risk IVDs, companion diagnostics, and IVDR implementation.
Companion diagnostic studies: BSI & TÜV SÜD Lead the Way in CDx Certification
June 6, 2023
The field of personalized medicine is experiencing notable progress as BSI and TÜV SÜD, two of the largest Notified Bodies in the European Union, have issued their first Companion Diagnostic (CDx) Certificates under the In Vitro Diagnostics Regulation (IVDR). This achievement carries substantial implications for manufacturers of in vitro diagnostics (IVD) and Contract Research Organizations (CRO) such as MDx CRO. MDx is a specialized IVD CRO with expertise in conducting clinical performance studies, including companion diagnostic studies, as stakeholders adapt to the changing landscape of CDx.
BSI The Netherlands (2797) issued its first CDx certificate in May, 2023, to Invivoscribe, Inc. for their LeukoStrat® CDx FLT3 Mutation Assay, a vital tool in tailoring treatment for acute myelogenous leukaemia (AML) patients with FLT3 ITD and TKD gene mutations.
Earlier, TÜV SÜD Product Service GmbH had made its mark as the issuer of the world’s first CDx certificate in accordance with the IVDR, awarded to Roche Diagnostics GmbH for a qualitative immunohistochemical cancer biomarker assay. This assay detects the programmed death-ligand 1 (PDL1) expression pattern, enabling identification of patients who will benefit most from a specific therapeutic treatment.
The milestones reached by Invivoscribe, Inc. and Roche Diagnostics GmbH signal to other manufacturers the effectiveness of the IVDR regulatory framework in certifying these devices. Furthermore, it testifies to the successful collaboration between manufacturers, EMA, and Notified Bodies, paving the way for similar certifications in the future.
Understanding the Impact of IVDR on Companion Diagnostics
Companion diagnostic devices like these are key in advancing personalized medicine, as they are clinically validated to determine patients’ likelihood of responding to a specific treatment. However, the enforcement of the IVDR has increased the regulatory oversight for these devices, pushing most into risk Class C, necessitating Notified Body review prior to being placed on the market.
The IVDR’s new risk classification concept has expanded the role of Notified Bodies like BSI and TÜV SÜD, requiring them to oversee more than 80% of IVD devices, a significant rise from the previous 10-15% under the IVD Directive. These regulatory changes underscore the value of experienced CROs like MDx CRO in supporting manufacturers through this intricate process.
Under the IVDR, CDx products, once free from Notified Body involvement, are now classified as Class C and must undergo a Notified Body conformity assessment. This process requires consultation with the European Medicines Agency (EMA) or the Competent Authority (CA) for medicinal products, as per the 2001/83/EC directive, which lengthens the overall conformity assessment process. Manufacturers must factor in this increased timeline, especially with the IVDR requiring Class C CDx products to be CE-marked by May 2026.
Choose MDx CRO for Reliable Companion Diagnostic Studies
As a trusted IVD CRO partner, MDx, with its extensive experience in managing IVD clinical performance studies including companion diagnostic studies, is equipped to assist clients through the evolving CDx and regulatory landscape. By optimizing innovative diagnostic solutions and ensuring full regulatory compliance in our CDx clinical trials, we strive to contribute to the delivery of top-tier personalized medicine, enhancing patient care worldwide.
Contact us today to learn how MDx CRO can partner with you to help bring your companion diagnostic to market.
IVD Software Development: How to Bring IVD Software to Market in 8 Steps
April 4, 2023
The healthcare industry is undergoing a rapid transformation spurred by the advent of advanced medical diagnostic technology. IVD software development is a critical component of this revolution as it allows for testing, analysis, reporting, and communication without needing a physical laboratory or a visit to a doctor’s office.
Bringing IVD software development to the market can benefit patients and healthcare providers who can deliver quality care faster with fewer resources.
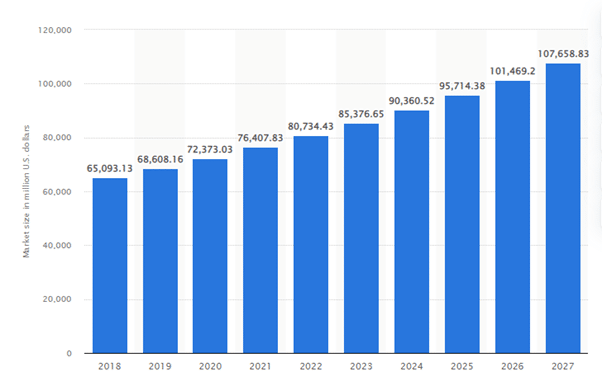
Projected size of the IVD market worldwide from 2018 to 2027 (in million U.S. dollars)
The above graph shows the In-Vitro Diagnostics (IVD) market globally was estimated at 72.4 billion U.S. dollars in 2020, with a projected growth of 108 billion U.S. Dollars by 2027, showing its increased relevance in the healthcare industry today.
1. Conduct Market Research
Before starting the development process, organizations must conduct market research and understand their target market, user needs, and potential competitors.
Market research should also help determine regulatory requirements that organizations must comply with and any current trends within the industry or technology space.
Below are some key points to consider when conducting market research for IVD software development:
Identify regulatory requirements: Identifying the regulatory requirements for your software is essential for bringing your product to market. They may vary depending on your target market, such as the FDA in the US or the European Union’s CE marking requirements (IVDR 746/2017).
Determine your target market: Identify the segments of the healthcare industry that your IVD software will serve. Consider factors such as geography, type of healthcare organization, and specialty areas.
Identify your competitors: Research the IVD software market to identify your competitors and their products. Analyze their strengths, weaknesses, pricing, and marketing strategies.
Understand your customers: Conduct surveys, interviews, and focus groups with healthcare professionals to understand their needs, preferences, and pain points. Use this information to tailor your IVD software to meet their specific needs.
Analyze market trends: Stay up-to-date on the latest trends and developments in the IVD software market. Monitor industry publications, attend conferences, and follow industry experts and thought leaders on social media.
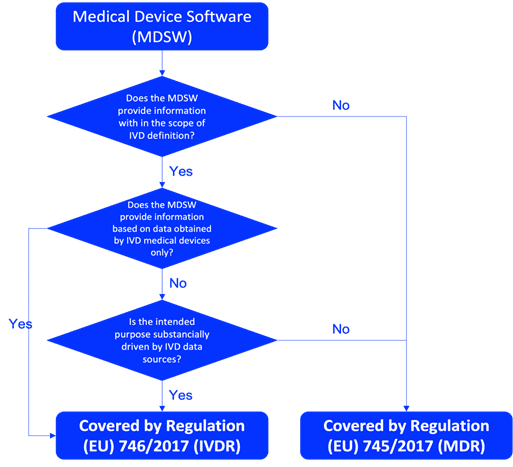
Understanding the classification of your IVD software is crucial before starting the planning and design process. Determine whether the IVD software is a standalone IVD medical device or a component of a larger system. To be qualified as an IVD medical device software in the EU, the product must fulfil the definition of IVD according to Article 2(2) of Regulation IVDR746/2017, as described in the Figure 1. The correct classification of IVD software should be done based on the rules described in Annex VIII of the IVDR. Relevant guidance documents, such as MDCG 2019-11 are also essentials for the qualification and classification of IVD software.
Fig. 1 – MDCG 2019-11 flowchart on qualification of Medical Device Software (MDSW)
Familiarize yourself with relevant regulatory frameworks, guidances and standards such as ISO 13485, IEC 62304, but also specific guidance documents published by regulators, which provide specifications and guidelines for developing, validating, and maintaining IVD software.
3. Plan and Design the Software
The next crucial step of a successful IVD software development is design and planning.
A well-documented and robust planning process can help provide a more detailed roadmap for development.
During this phase, design reviews, testing, and verification will ensure that the final version meets user requirements.
It is essential to incorporate user feedback at every stage of the design process to develop an intuitive interface that works effectively according to their needs.
Obtaining feedback from customers and stakeholders offers the development team opportunities to recognize potential concerns and areas for enhancement.
Developers can devise software solutions that fulfill customer needs and address their grievances by integrating feedback.
The importance of accurate documentation should not be underestimated as it helps trace back issues later on in the lifecycle of the software.
The development team must consider scalability and flexibility during the initial planning and design stages when creating the software.
4. Develop and Test the Software
Developing and testing the software is crucial in creating a working prototype.
The development phase is necessary to ensure the accuracy of the design, coding, and algorithms used in creating the software solutions.
Testing and quality assurance also play an essential role in ensuring the products meet all requirements before launch.
It is essential for companies to thoroughly assess each component of their software as part of this process. This includes ensuring they meet performance objectives concerning speed, responsiveness, scalability, security, reliability, and ease of use for their users.
Quality assurance checks help identify bugs or errors to release a defect-free product that meets all standards from regulatory bodies such as FDA or CE Marking.
When deploying IVD systems, manufacturers need to consider if their applications can be flexible enough to support new technological advances; future-proofing their products becomes increasingly necessary where customers demand longevity across upgrades or iterations over time.
5. Prepare a Regulatory Submission
Preparing a regulatory submission package is critical in bringing IVD software development to market. This step involves compiling documents to demonstrate that the software meets regulatory requirements and is safe and effective.
Here are some critical considerations for preparing a regulatory submission:
Gather relevant documents for the package
Understand regulations, standards, and risk classification of IVD software and the manufacturing role. Key documents to include are the device description, technical documentation, risk management file (ISO 14971), software lifecycle documentation (IEC 62304), and quality management system documentation (ISO 13485). Risk management must be applied and monitored during the IVD software development life cycle.
Prepare a performance evaluation report (PER)
This will require a comprehensive analysis of scientific evidence showing that the product meets user needs safely and effectively. IVD software performance evaluation should be prepared in accordance with relevant guidance documents, such as MDCG 2020-1 for the EU. Other guidance such as MedTech Europe Clinical Evidence Requirements for IVD can also be a good source of additional information.
Clinical performance studies are aimed at providing evidence of the safety and effectiveness of a product’s intended purpose to ensure that it’s able to diagnose, monitor and predict diseases and conditions accurately.
As described in MDCG 2020-1“Validation of the clinical performance should be considered at each change of the software to a new release. If no validation is performed, a justification should be stated in the technical documentation. With a validation of clinical performance, it is demonstrated that users can achieve clinically relevant outputs through predictable and reliable use of the MDSW”.
Adherence to relevant standards and guidelines, such as ISO 20916 (Clinical Performance Studies for In vitro diagnostics) and Good Clinical Practice (CGP), are crucial for the successful execution of clinical studies.
Ensure data accuracy
Ensure that that any data collected from testing is presented accurately to prove safety and efficacy before submitting your application. This includes validation and verification data, performance evaluations, and, if applicable, results from clinical studies. Carefully review all information for accuracy and completeness before submitting it.
6. Obtain Regulatory Approval
You need to obtain regulatory approval to bring IVD software development to market. Successful approval enables you to market and sell your software in compliance with local laws and regulations.
Familiarize yourself with the applicable regulatory frameworks and guidelines for your software. These may include ISO 13485 (quality management systems), IEC 62304 (medical device software lifecycle processes), ISO 14971 (risk management), and MDCG guidance documents such as MDCG 2019-11 (qualification and classification of software in the medical device regulations) and MDCG 2020-1 (guidance on performance evaluation for IVD software under the IVDR. You should also consider guidelines from other jurisdictions depending on your market strategy. The FDA for example has issued guidance for software as a medical device (SAMD).
Develop a comprehensive application package that should include all the necessary documents, data, and tests required for review. Ensure your package addresses regulatory requirements, such as conformity assessment procedures, clinical evidence, and post-market surveillance.
Submit the application package to the relevant regulatory authorities for review, such as the FDA in the US or notified bodies in the EU under the In Vitro Diagnostic Regulation (IVDR) 2017/746.
Be prepared to respond quickly and accurately to any feedback or additional information requests from regulatory agencies during the review process.
Make recommended changes swiftly as part of your submission to obtain approval from agencies successfully.
Once you receive approval from regulatory agencies, you can move forward with marketing and selling your software according to local laws & regulations, ensuring ongoing compliance with any post-market requirements.
7. Developing Marketing and Sales Strategies
Creating a successful marketing and sales strategy is essential for bringing IVD software to the market, it allows for faster positioning and gaining a competitive advantage. Make sure to develop a strong brand identity with messaging that resonates with your audience.
In addition, researching customer needs and understanding key industry trends can create a more targeted approach when it comes to the marketing of IVD software solutions, increasing your likelihood for success.
Make sure to use multiple channels such as paid advertising, email campaigns, social media and webinars to reach out to potential customers from diverse segments.
And last but not least, creating effective communication strategies to engage with customers throughout the sales cycle will also be key to promoting IVD products successfully.
8. Launch and Support the Software
Launching and supporting software is a crucial element to its success. The product can be improved over time by providing regular updates and customer service, and users can get the best experience.
Here are some points to consider when launching your IVD software development:
Create a comprehensive support plan that puts customer needs first. Ensure you have an efficient process for handling inquiries and technical issues as they arise.
Ensure that all necessary software updates are completed on schedule, so users don’t experience any delay in accessing the product’s full features or bug fixes.
Assess the regulatory impact of changes and bug fixes, changes to your IVD software may or may not be significant. Consider whether changes impact your regulatory approvals. MDCG 2022-6 provides additional guidance on changes to design and intended purpose in the context of the new transition timelines for IVDs in Europe.
Set up user feedback forms or surveys so customers can share their thoughts on the product’s performance and what improvements they want to see. This will help drive further development of the software over time.
Offer ongoing training opportunities for new features, so users feel confident using them once released. This will also ensure that customers know how to use their investment in your IVD software development solution fully.
Revolutionizing Healthcare With IVD Software Development
In vitro diagnostic (IVD) software development has transformed the healthcare industry by providing cost-effective testing, analysis, reporting, and communication solutions without physical laboratory equipment.
Studies indicate that healthcare providers highly prioritize in vitro diagnostic (IVD) procedures, and their optimization has the potential to enhance patient outcomes. Therefore, the development of IVD software is crucial in facilitating quicker and more accurate diagnostic results, ultimately leading to the optimization of healthcare practices.
If you need a partner in IVD software development for your business, MDx CRO is an IVD consultancy that provides end-to-end solutions to accompany you at each step of the process. Our team of highly experienced CRO strategists has extensive expertise in bringing innovative medical devices and IVD technologies to market. Request your expert consultation today.
FAQs
What are the key considerations when designing IVD software?
There are several key considerations that companies should keep in mind when designing IVD software: user requirements, regulatory requirements depending on the target geographic location, data accuracy and effective data management, the software’s ability to integrate with other systems, as well as performance and usability.
What are the regulatory requirements for IVD software development in Europe?
The regulatory requirements for IVD software development in Europe are determined by the In Vitro Diagnostic Regulation (IVDR), which became applicable on May 26, 2022. They include, but are not limited to design and development, risk management, validation and verification, as well as compliance with GDPR.
What are the most common challenges in IVD software development?
The most common challenges in IVD software development include regulatory compliance (which can be complex and challenging to navigate through), ensuring integration compatibility with other systems, effective data management, and great user experience, among others.
How do you ensure the quality and reliability of IVD software?
To ensure the quality and reliability of IVD software, it’s important that companies follow all regulatory guidelines applicable to their geographical location, and use a quality management system to ensure that the development process is well-documented. Conducting testing, validation and verification processes is another essential element of software development for in vitro diagnostics.
Gain access to MDx’s Precision Medicine Services Pack—detailing our capabilities in clinical research, regulatory strategy, and companion diagnostics. Learn how we support pharma, CDx, and advanced therapy developers across the clinical and regulatory lifecycle.