
Esta guía sobre el estudio de rendimiento clínico del anexo XIV del IVDR explica cómo puede planificar y obtener la autorización para estudios de rendimiento en virtud del anexo XIV del IVDR cuando implica un diagnóstico complementario. Su objetivo es ser práctica y estar alineada con las expectativas actuales de los comités de ética y las autoridades competentes en la Unión Europea.
Si necesita una lista de verificación estructurada, puede descargar el kit de herramientas de autorización de estudios de rendimiento del anexo XIV (PSA), que incluye una lista de verificación de monitorización ISO 20916, plantillas y un plan de trabajo previo a la presentación.
Autorizado para el reclutamiento: guía práctica para estudios del anexo XIV del IVDR
Descargue el white paper aquí.
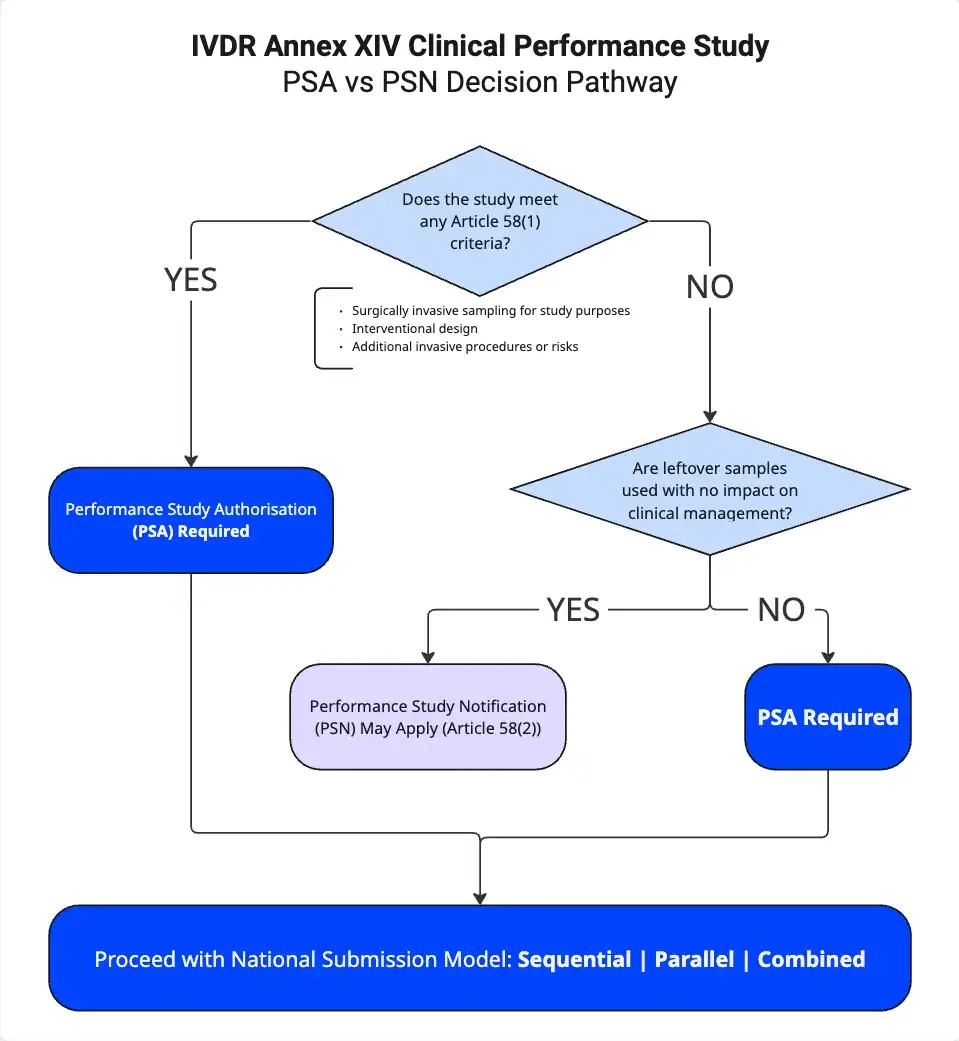
Estudio de rendimiento clínico del anexo XIV del IVDR: cuándo se aplica PSA vs. PSN para diagnósticos complementarios
Los diagnósticos complementarios (CDx) suelen requerir un estudio de rendimiento clínico del anexo XIV del IVDR porque estas pruebas influyen directamente en el manejo del paciente. Por ello, comprender cuándo se aplica una autorización de estudio de rendimiento (PSA) o una notificación de estudio de rendimiento (PSN) es fundamental para una planificación regulatoria eficaz y para evitar retrasos innecesarios.
Por qué los diagnósticos complementarios suelen quedar bajo el anexo XIV
Los diagnósticos complementarios suelen quedar bajo el anexo XIV del IVDR porque el resultado de la prueba guía decisiones clave de tratamiento, entre ellas:
- Selección de pacientes
- Asignación del tratamiento
- Continuación o interrupción de la terapia
Si el diseño del estudio permite que los resultados de la prueba influyan en las decisiones clínicas, los reguladores consideran que el estudio es intervencionista, y esta clasificación activa la necesidad de una autorización de estudio de rendimiento (PSA). Además, si utiliza el dispositivo fuera de su finalidad prevista tal como se define en las instrucciones de uso (IFU), el marco del IVDR también exige una PSA.
¿Cuándo se aplica una PSA en virtud del artículo 58(1)?
Para cualquier estudio de rendimiento clínico del anexo XIV del IVDR, el artículo 58(1) es la disposición clave para determinar si se requiere una PSA. Una PSA pasa a ser obligatoria si se cumple cualquiera de los tres criterios siguientes:
- Realiza una toma de muestras quirúrgicamente invasiva específicamente para el estudio de rendimiento clínico (CPS).
- Diseña el estudio con un carácter intervencionista.
- Introduce procedimientos invasivos adicionales u otros riesgos para los participantes.
Si se aplica aunque sea uno de estos criterios, debe obtener una autorización de estudio de rendimiento antes de iniciar el estudio.
¿Cuándo se aplica una PSN en virtud del artículo 58(2)?
Si no cumple ninguno de los criterios del artículo 58(1), puede aplicarse en su lugar el artículo 58(2). En ese caso, puede presentar una notificación de estudio de rendimiento (PSN) cuando:
- El estudio utiliza únicamente muestras sobrantes
- El estudio no incluye procedimientos invasivos adicionales
- Los resultados de la prueba no influyen en el manejo del paciente
- El diseño sigue siendo estrictamente no intervencionista
No obstante, incluso cuando se cumplen estas condiciones, debe evaluar cuidadosamente los requisitos nacionales y los detalles específicos del diseño del estudio para confirmar que una PSN sigue siendo adecuada.
Estudios combinados de medicamento y diagnóstico
Un estudio de rendimiento clínico del anexo XIV del IVDR que implique tanto un diagnóstico complementario como un medicamento requiere una coordinación estructurada desde el inicio. Cuando se aplican ambos marcos regulatorios, el Reglamento de Ensayos Clínicos (CTR) rige el medicamento, mientras que el IVDR rige el diagnóstico. En consecuencia, debe alinear los plazos, la documentación y la estrategia regulatoria en ambos marcos para evitar incoherencias y retrasos.
Paso a paso: de la planificación a la aprobación de la PSA
1. Elementos esenciales del IB y el CPSP, y el vínculo entre criterios de valoración, finalidad prevista y estrategia de punto de corte
Comience con un folleto del investigador (IB) y un plan del estudio de rendimiento clínico (CPSP) coherentes. Cada afirmación del CPSP debe poder trazarse hasta la finalidad prevista del dispositivo y hasta la evidencia analítica y clínica suficiente para sustentarla. Los criterios de valoración deben alinearse con la decisión clínica prevista.
Según nuestra experiencia en más de 100 proyectos, los dos hallazgos de revisión siguientes se repiten con frecuencia:
- Punto de corte analítico y validación. Las autoridades examinan de cerca cómo se ha definido y sustentado el punto de corte del ensayo. La sensibilidad, la precisión y la exactitud deben demostrar que el dispositivo funciona de un modo que respalda decisiones clínicas seguras. Una justificación débil invita a preguntas sobre el riesgo de clasificación errónea de pacientes, lo que con frecuencia conduce a solicitudes de información.
- Criterios de valoración no alineados con la finalidad prevista. Los revisores cuestionan con frecuencia criterios de valoración primarios que no están claramente vinculados al rendimiento clínico o a la finalidad prevista del dispositivo. El criterio de valoración debe corresponder directamente a la decisión que se toma para el paciente.
Medidas prácticas que hemos implementado en más de 100 proyectos:
- Elabore el plan de análisis estadístico de forma temprana y muestre una línea clara desde la finalidad prevista hasta el criterio de valoración y los criterios de éxito.
- Vincule cada estudio analítico (límite de detección, límite de cuantificación, precisión, interferencias, justificación del punto de corte) con la afirmación clínica que respalda.
2. Presentaciones por país: comités de ética, autoridades competentes, portales, traducciones y tasas
Planifique tanto la vía del comité de ética como la de la autoridad competente, incluidas las cuentas para portales nacionales, las políticas de traducción y el pago de tasas. Los requisitos específicos de cada país pueden modificar los plazos y la logística.
Algunos ejemplos de requisitos específicos por país:
- Francia requiere un código de registro IDRCB. El protocolo, el formulario de consentimiento informado y el certificado de seguro deben mostrar este código. La ausencia del código o su uso incoherente suele desencadenar solicitudes de información.
- Polonia puede requerir un paquete de presentación físico en lugar de un expediente totalmente electrónico. Planifique en consecuencia todos los documentos que requieran firmas originales. El tiempo de mensajería, las copias notarizadas cuando proceda y la secuenciación de firmas también deben incorporarse al calendario.
Una breve lista de verificación previa a la presentación:
- Identificación del enfoque de presentación CE-ACN por país (secuencial, paralelo, combinado) y calendarios de reuniones del CE
- Acceso al portal verificado y roles asignados tanto para comités de ética como para autoridades competentes.
- Identificadores nacionales obtenidos y propagados de forma coherente en todos los documentos.
- Alcance de la traducción definido y con control de calidad, especialmente para materiales dirigidos a pacientes.
- Certificados de seguro alineados con el alcance del estudio y las expectativas nacionales.
- Tablas de tasas confirmadas y órdenes de compra preparadas.
3. Plazos, paradas de reloj y consultas de expertos
Las fases de validación y evaluación de la revisión suelen incluir paradas de reloj para aclaraciones, en las que la autoridad revisora puede solicitar información adicional, conocidas como Request for Informations (RFIs). Debe definir con antelación los niveles de servicio internos para las respuestas, y la titularidad de los temas debe estar clara entre los contribuyentes analíticos, clínicos y de bioestadística. Una referencia cruzada maestra que vincule CPSP, IB, gestión de riesgos y secciones estadísticas reduce el riesgo de respuestas incoherentes. En la evaluación de Pickett, asignar la titularidad de los temas para las respuestas analíticas, clínicas y estadísticas antes de la presentación ayuda a mantener cortas las paradas de reloj y evita respuestas incoherentes entre documentos.
Estudio de rendimiento clínico del anexo XIV del IVDR: ¿cómo elaborar un expediente sólido?
Un expediente sólido de estudio de rendimiento clínico del anexo XIV del IVDR reduce el riesgo de solicitudes de información (RFIs), paradas de reloj y retrasos en la aprobación.
Validación analítica y justificación del punto de corte
Para un estudio de rendimiento clínico del anexo XIV del IVDR, la justificación del punto de corte debe ir más allá de presentar un único valor umbral o una curva ROC.
Un expediente sólido debería:
- Explicar las consecuencias clínicas del punto de corte seleccionado
- Describir cómo cambian la sensibilidad y la especificidad si se desplaza el umbral
- Abordar los falsos positivos y falsos negativos con una prevalencia clínicamente relevante
- Vincular el rendimiento analítico directamente con el criterio de valoración primario
- Demostrar cómo el dispositivo respalda una decisión clínica segura
Las autoridades se centran con frecuencia en el riesgo de clasificación errónea. Si la justificación del punto de corte no respalda claramente una toma de decisiones segura, esta sección se convierte en un factor principal de RFIs.
Mejores prácticas según Callum Pickett, Clinical Alliance Lead en MDx
Trate la justificación del punto de corte con el mismo rigor que un argumento de seguridad. Aporte tanto evidencia estadística como una narrativa clínica clara.
Estrategia de consentimiento informado alineada con el CPSP
La falta de alineación entre el plan del estudio de rendimiento clínico (CPSP) y el formulario de consentimiento informado es una causa frecuente de retraso en un estudio de rendimiento clínico del anexo XIV del IVDR.
Para reducir el riesgo:
- Finalice primero el CPSP.
- Redacte el consentimiento informado para reflejar los procedimientos, los calendarios de visitas y los riesgos.
- Utilice un lenguaje claro y sencillo que refleje con precisión el protocolo.
Los revisores evalúan si los participantes están debidamente informados. Si el documento de consentimiento no refleja el diseño del estudio, es probable que haya un RFI.
Recomendamos incluir lo siguiente en el consentimiento informado:
- Resumen claro de los procedimientos y del calendario de evaluaciones
- Riesgos específicos del dispositivo, incluida la manipulación de muestras y la posible repetición de pruebas
- Explicación de resultados inválidos o indeterminados y sus implicaciones para el participante
La coherencia entre el CPSP y la documentación de consentimiento refuerza la credibilidad durante la evaluación.
Referencias cruzadas: CPSP, IB, GSPR e informes del estudio
Una matriz de referencias cruzadas mejora tanto el control de calidad interno como la eficiencia de la revisión externa.
Su matriz debería demostrar:
- Dónde se aborda cada requisito general de seguridad y rendimiento (GSPR)
- Cómo se monitorizan y documentan los procedimientos del CPSP
- Dónde se sustentan los compromisos estadísticos mediante análisis
- Cómo la gestión de riesgos se vincula con los controles del estudio
Para una presentación exitosa de un estudio de rendimiento clínico del anexo XIV del IVDR, la trazabilidad documental es fundamental.
Factores frecuentes que impulsan RFIs en estudios de rendimiento clínico del anexo XIV del IVDR
A continuación se indican deficiencias habituales y estrategias prácticas de mitigación:
| Factor que impulsa el RFI | Cómo abordarlo |
|---|---|
| Criterio de valoración primario no alineado con la finalidad prevista | Redefina o reformule el criterio de valoración para que respalde directamente la decisión clínica |
| Justificación del punto de corte insuficiente | Aporte datos analíticos completos y explique el impacto clínico |
| Representatividad de la muestra poco clara | Justifique el tipo de matriz, el estadio de la enfermedad, la terapia previa y las variables relevantes |
| Clasificación errónea del tipo de estudio (muestras sobrantes) | Aclare si el diseño sigue siendo no intervencionista y si la PSN es adecuada |
| Plan de monitorización no alineado con ISO 20916 | Defina categorías de acontecimientos adversos, roles y plazos |
| Consentimiento informado incoherente con el CPSP | Alinee con precisión el lenguaje y los detalles procedimentales |
| Supuestos estadísticos no justificados clínicamente | Vincule alfa y potencia a diferencias clínicas significativas |
| Notificación de deficiencias del dispositivo poco clara | Defina mecanismos de detección, escalado y notificación |
| Gestión de riesgos no conectada con los controles del estudio | Trace los riesgos hasta las actividades de mitigación y monitorización |
| Gobernanza combinada CTR–IVDR poco clara | Defina roles, responsabilidades y vías de decisión |
| Traducciones incompletas o de baja calidad | Planifique revisión profesional y retrotraducción |
| Identificadores nacionales o seguro no coinciden | Asegure códigos coherentes y límites de cobertura adecuados |
Guía por país: Cómo planificar solicitudes de estudios de rendimiento a Estados miembros de la UE
1. Identifique el enfoque de presentación de la PSA adoptado por el Estado miembro de la UE.
Existen tres modelos adoptados por los países de la UE que afectarán a su estrategia de presentación:
- Secuencial – se presenta primero al CE y la presentación a la ACN solo puede realizarse una vez emitida la aprobación del CE.
- Paralelo – las presentaciones al CE y a la ACN pueden presentarse alrededor del mismo momento o al mismo tiempo, lo que permite un proceso de revisión “paralelo”. No obstante, la ACN solo aprobará el estudio una vez se haya concedido una opinión favorable del CE
- Combinado – se envía una única presentación de PSA a una autoridad que actúa como CE y ACN; se emitirá una única opinión favorable.
2. Elija su comité de ética:
- Se recomienda encarecidamente presentar la PSA al mismo CE que revisa la solicitud de ensayo clínico asociada
- Identifique cualquier plantilla específica del CE; esto puede incluir formularios de solicitud específicos del CE y plantillas de documentos del centro
- Identifique el calendario de reuniones del CE y los plazos para la presentación de la PSA a fin de lograr la revisión en la fecha de la reunión del CE
- Utilice el calendario de reuniones del CE para informar su estrategia de presentación; distintos CE se reunirán con frecuencias diferentes.
3. Identifique cualquier requisito específico establecido por la autoridad nacional competente:
- ¿Existen plantillas específicas de la ACN que deban presentarse con la PSA?
- ¿Hay algo que pueda condicionar la presentación en este país? Por ejemplo, ¿la ACN exige que el protocolo final del ensayo clínico se presente junto con la presentación?
4. Identifique cualquier ley y requisito específico del Estado miembro de la UE al que se presenta:
- La UE se rige por las leyes del RGPD, pero las leyes nacionales de protección de datos añaden una capa adicional de requisitos. Asegúrese de que su estudio se desarrolla teniendo en cuenta los requisitos nacionales de protección de datos.
- Los Estados miembros de la UE tienen requisitos diferentes para la documentación del seguro; esto puede incluir referencias a leyes nacionales, la inclusión de códigos nacionales del estudio y detalles adicionales sobre el número de participantes.
5. Utilice todos estos puntos para crear su estrategia de presentación, informada por lo siguiente:
- Enfoque de presentación: los países con enfoques de revisión secuencial tardan, de media, más que los países con enfoques de revisión paralela y combinada.
- Clinical Priority: what countries are priority for enrolment? Which countries will have the most sites and therefore need to be activated earlier?
- ¿Con qué frecuencia se reúnen los CE elegidos según su calendario de reuniones?
- ¿Hay requisitos documentales de la ACN o del CE que aún no estén disponibles y puedan retrasar la presentación?
Monitorización conforme a ISO 20916
ISO 20916 introduce categorías de clasificación adicionales para acontecimientos adversos en comparación con el texto base del IVDR. Los centros necesitan una formación clara sobre la taxonomía de eventos, las responsabilidades de clasificación y los plazos de notificación.
Contenido que debe incluirse en el plan de monitorización y en la formación del centro:
- Definiciones y ejemplos de acontecimientos adversos y acontecimientos adversos graves utilizados en el estudio.
- Roles para la clasificación inicial, la revisión médica y la evaluación final.
- Plazos específicos de notificación desde el centro al promotor y del promotor a las autoridades.
- Cómo capturar, evaluar y notificar las deficiencias del dispositivo.
Tal como ha observado nuestro equipo clínico en presentaciones del anexo XIV, la formación temprana sobre la taxonomía de acontecimientos adversos y los plazos de notificación es esencial. La clasificación errónea en el primer caso notificado suele dar lugar a acciones correctivas y a impacto en el calendario.
Validez científica dentro de la evaluación del rendimiento del anexo XIV
De la PSA al mercado: coordinación con el organismo notificado y los reguladores de medicamentos
Para una aprobación real de diagnóstico complementario, alinee la validez analítica, el rendimiento clínico y la validez científica con los planes poscomercialización. El lenguaje del etiquetado y las expectativas de evidencia deben coordinarse con el organismo notificado y, cuando proceda, con los reguladores de medicamentos. Planifique la transición desde la evidencia del estudio al seguimiento del rendimiento poscomercialización.
Según Callum Pickett, mantener un único mapa de evidencia que vincule la validez analítica, el rendimiento clínico y la validez científica con el lenguaje final del etiquetado agiliza la revisión del organismo notificado y reduce las rondas de aclaración posteriores a la presentación.
Recursos
- Orientación del Medical Device Coordination Group (MDCG) y de la Comisión Europea sobre estudios de rendimiento, incluida una sección de preguntas y respuestas sobre las vías del artículo 58.
- Orientación nacional, como la de la Agencia Federal Belga de Medicamentos y Productos Sanitarios (FAMHP), sobre la estructura del expediente, los plazos y las tasas para estudios de rendimiento.
- Visiones generales de consultoría, como DLRC Group (DLRC Group), para un contexto paneuropeo.
- Normas publicadas por la International Organization for Standardization (ISO), en particular ISO 20916 para estudios de rendimiento clínico de dispositivos médicos de diagnóstico in vitro.
Autorizado para el reclutamiento: guía práctica para estudios del anexo XIV del IVDR
Descargue el white paper aquí.
Perspectiva experta de Callum Pickett
El éxito con estudios del anexo XIV para diagnósticos complementarios depende de la alineación. La finalidad prevista, los criterios de valoración, la validación analítica y el punto de corte, el consentimiento y la monitorización deben ser coherentes y apoyarse mutuamente. La atención cuidadosa a los requisitos específicos por país y la planificación temprana de la coordinación CTR-IVDR reducen la probabilidad de paradas de reloj y solicitudes de información. Una lista de verificación estructurada y unas referencias cruzadas disciplinadas mejoran la calidad del expediente y la eficiencia de la evaluación.
Preguntas frecuentes (FAQ)
Se requiere una PSA en virtud del artículo 58(1) si el estudio incluye una toma de muestras quirúrgicamente invasiva específicamente para el estudio, utiliza un diseño intervencionista en el que los resultados de la prueba influyen en el manejo del paciente, o introduce procedimientos invasivos adicionales o riesgos. En la práctica, los diagnósticos complementarios suelen activar una PSA porque sus resultados guían decisiones de tratamiento. Por lo tanto, en cuanto se aplique uno de estos criterios, debe obtener una PSA antes de iniciar el estudio. Para un contexto más amplio sobre la realización de estudios bajo el IVDR, lea el siguiente artículo sobre la realización de estudios clínicos bajo el IVDR
Puede utilizar una PSN en virtud del artículo 58(2) cuando el estudio siga siendo estrictamente no intervencionista. Por ejemplo, el estudio puede basarse únicamente en muestras sobrantes, evitar procedimientos invasivos adicionales y garantizar que los resultados de la prueba no influyan en las decisiones clínicas. No obstante, debe evaluar el diseño cuidadosamente, porque clasificar erróneamente un estudio como no intervencionista con frecuencia provoca retrasos y solicitudes de reclasificación.
En estudios combinados, debe cumplir simultáneamente ambos marcos: el CTR rige el medicamento, mientras que el IVDR rige el diagnóstico complementario. Como resultado, debe alinear desde el inicio los criterios de valoración, la finalidad prevista y la población de pacientes en ambas presentaciones. De lo contrario, las incoherencias entre los expedientes del CTR y del IVDR suelen desencadenar solicitudes de información y paradas de reloj. Un análisis de brechas estructurado puede ayudarle a identificar y resolver estos riesgos de forma temprana. Lea aquí el artículo sobre la evaluación previa a la presentación.
Las autoridades suelen emitir RFIs cuando los promotores no alinean los criterios de valoración primarios con la finalidad prevista, aportan una validación analítica o una justificación del punto de corte insuficientes, o no explican claramente el riesgo de clasificación errónea. Además, las incoherencias entre el CPSP y el consentimiento informado, o una trazabilidad débil entre la gestión de riesgos y los supuestos estadísticos, suelen generar preocupaciones. Por ello, debe construir una estructura clara de referencias cruzadas en todos los documentos para reducir fricciones en la revisión.
Para más detalles sobre las expectativas de documentación, lea la documentación técnica de IVD.
Para reducir los plazos, debe definir los criterios de valoración de forma temprana y vincularlos directamente con la finalidad prevista, justificar los puntos de corte analíticos con evidencia estadística y fundamento clínico, y alinear el consentimiento informado con precisión con el CPSP. Al mismo tiempo, confirme antes de la presentación los modelos nacionales de presentación, los calendarios de los comités de ética, el alcance de la traducción y los requisitos de seguro. Al asignar una titularidad interna clara para las respuestas analíticas, clínicas y estadísticas, también puede acortar las paradas de reloj y mantener la coherencia durante la revisión.







