FDA’s new LDT regulation underscores the vital role of IVD CROs in compliance and safety. Phased over four years without grandfathering, it highlights the necessity for IVD CROs’ regulatory expertise and their key role in aligning LDTs with stringent standards, marking a significant shift in the IVD landscape.
Introduction
The Food and Drug Administration (FDA) recently released a groundbreaking proposed rule on September 29, 2023, aiming to transform its existing approach toward Laboratory-Developed Tests (LDTs). This paradigm shift directly impacts In Vitro Diagnostic products (IVDs) and their regulation. The proposed rule presents a layered plan for LDTs, phasing out FDA’s longstanding policy of enforcement discretion. For stakeholders in the IVD sector, including clinical laboratories and LDT manufacturers, the implications of this proposed rule are monumental.
Key Points to Consider as the FDA regulates LDTs
Modification to IVD Definition: The FDA aims to explicitly categorize LDTs as IVDs within the scope of 21 CFR Part 809.3, thus making them subject to FDA’s medical device regulations, including premarket review.
Phased Implementation: The FDA proposes to gradually roll back its enforcement discretion policy for LDTs, segmenting the regulatory shifts over five distinct stages across a four-year period.
No Grandfather Clause: Unlike previous considerations, the new proposal doesn’t plan to “grandfather” existing LDTs. Public commentary on this subject is invited.
Test Exemptions: Specific test types, such as forensic and human leukocyte antigen tests, are marked for exemption from enhanced regulatory oversight.
Comment Period: Stakeholders have until December 4, 2023, to submit their feedback on the proposed rule.
Background on FDA Regulation for LDTs and IVDs
IVDs have traditionally been subject to rigorous regulatory scrutiny under various heads:
510(k) premarket notification or premarket approval (PMA)
Quality system regulation
Medical device reporting
Registration and listing
Labeling
These IVDs also fall under the Clinical Laboratory Improvement Amendments of 1988 (CLIA). LDTs, a specialized category within IVDs, have long operated under enforcement discretion, essentially receiving less stringent oversight. This approach historically aligned with the perception of LDTs as low-risk products, but the proposed rule acknowledges the evolved complexity and widespread use of LDTs in modern healthcare.
The Evolving Landscape of LDTs
Over the past 50 years, the role of LDTs has changed dramatically, making the FDA regulation of Laboratory Developed Tests increasingly crucial. Their growing prevalence and the technical sophistication involved have prompted calls for stronger regulatory oversight. These significant developments have culminated in the FDA’s renewed perspective on LDTs. Shedding its previous stance of general enforcement discretion, the FDA Laboratory Developed Tests Regulation now aims to redefine LDTs broadly, addressing potential regulatory gaps and emphasizing the need for robust public health protection.
The Road Ahead: Implications and Recommendations
The phased implementation approach impacts various aspects of medical device regulation:
Medical Device Reporting: Will be the first area where enforcement discretion will cease.
Quality Systems: Expected to come under scrutiny three years after the final policy is published.
Premarket Review: To be phased in 3.5 to four years after the final policy, impacting high-risk IVDs first and then trickling down to moderate-risk and low-risk IVDs.
Given these imminent changes, clinical labs offering LDTs must prepare for enhanced regulatory compliance and for increased FDA regulation of Laboratory developed tests. These labs should develop protocols for both analytical and clinical validity, ensuring alignment with new regulatory expectations.
Synergies with the EU’s IVDR roll-out
In Europe, a similar regulation, the In Vitro Diagnostic Regulation (IVDR), is being phased in, emphasizing the importance of compliance for in house developed tests. Under IVDR, laboratories must ensure that their Technical Documentation and Quality Management System are up-to-date and comply with the regulation and the national law. Failure to comply with the IVDR can result in serious consequences, including fines, loss of accreditation, and even closure of the laboratory.
Furthermore, laboratories that develop in-house IVDs will be required to comply with similar requirements for manufacturing as commercial IVD manufacturers. This means that laboratories must ensure that their IVDs meet high standards for safety and performance, which can only be achieved through rigorous testing and validation.
It is important to note that the IVDR specifically includes in-house developed tests, including LDTs and CE marked IVDs modified by laboratories. If an equivalent CE marked device is available on the market or IVD product is manufactured at an industrial scale, the laboratory cannot use exemption for LDT´s provided in the IVDR (Article 5.5) and must CE mark the IVD. MDx CRO has recently published an article assessing the impact of the IVDR on LDTs.
Conclusion: An Industry in Transition
As specialists in quality, regulatory, and clinical consultancy focused on IVDs, MDx CRO advises stakeholders to take proactive measures in anticipation of these sweeping regulatory changes. Judicial challenges and public feedback could alter the timeline for the FDA to start regulating LDTs, but there’s no doubt that enhanced regulation of LDTs is on the horizon, already in full force in Europe.
Comments on the Proposed FDA Rule should be submitted by December 4, 2023, to help shape this crucial regulatory development. It is an opportune time for the industry to engage in open dialogue and to prepare for the inevitable changes that lie ahead.
Written by:
Carlos Galamba
CEO
Senior regulatory leader and former BSI IVDR reviewer with deep experience in CE marking high-risk IVDs, companion diagnostics, and IVDR implementation.
MDx: Your Dedicated CRO for IVD Clinical Studies in the EU
August 24, 2023
Introduction
In the rapidly evolving world of in vitro diagnostics (IVD), manufacturers are increasingly understanding the need for rigorous clinical performance studies. Such studies form the backbone for ensuring the safety, efficiency, and overall market readiness of IVD devices. With the European Union’s (EU) stringent regulatory environment, conducting these studies requires expertise and precision. That’s where MDx CRO, a trusted name in IVD Contract Research and regulatory consulting, stands out.
Evidence-based Decision Making: Clinical performance studies furnish the data that can prove the diagnostic accuracy, precision, and utility of IVD devices. They help manufacturers refine their offerings and justify their product claims.
Regulatory Adherence: Ensuring compliance with the EU’s In Vitro Diagnostic Regulation (IVDR) and standards like ISO 20916 is non-negotiable. Clinical studies often form the bedrock in gaining these credentials and opening up the European market.
Navigating the Challenges with MDx CRO
Whether you’re a fledgling startup or an established IVD giant, challenges like site selection, study design, effective monitoring, and regulatory adherence can be daunting. This is where MDx CRO can be your guiding light:
Proven Expertise: With its legacy in the IVD realm and former Notified Body experts on board, MDx CRO offers unparalleled insights into effective study design, ensuring manufacturers derive actionable insights every time.
Network of Clinical Sites: Owing to its years in the industry, MDx CRO has built strong affiliations with leading clinical sites, guaranteeing timely and efficient study conduct.
Regulatory Insight: Navigating the IVDR and ISO 20916 maze becomes simpler with MDx CRO’s regulatory consulting wing, which ensures manufacturers always stay on the right side of the law.
End-to-End Monitoring: With a keen focus on detail, MDx CRO ensures every study stays on track, protocols are maintained, and data integrity remains uncompromised.
Why MDx CRO?
Simply put, MDx CRO isn’t just a service provider – it’s a partner in your IVD journey. Our seasoned team understands the unique challenges IVD manufacturers face, making them an indispensable asset in your product’s journey from concept to the European market.
Conclusion
IVD clinical studies, while challenging, present a golden opportunity to IVD manufacturers to rigorously validate their product’s claims. In the intricate web of EU regulations, manufacturers need more than just expertise; they need a partner. And who better than MDx CRO, which has consistently demonstrated excellence in study design, monitoring, and ensuring complete regulatory compliance? Choose MDx CRO, and let’s work together to bring transformative and reliable IVD devices to the EU market.
FAQs about IVD Clinical Studies and MDx CRO:
What are IVD Clinical Studies?
IVD clinical studies refer to rigorous research and evaluations conducted to determine the safety, efficiency, and overall performance of in vitro diagnostic (IVD) devices.
Why are IVD Clinical Studies important in the EU?
The EU has stringent regulatory requirements. IVD clinical studies provide the necessary evidence to support product claims, ensuring compliance with the EU’s In Vitro Diagnostic Regulation (IVDR) and international standards like ISO 20916.
What challenges can manufacturers expect while conducting IVD studies in the EU?
Manufacturers may face challenges like site selection, creating an effective study design, regular study monitoring, and ensuring compliance with EU regulations and standards.
How does MDx CRO help with these challenges?
MDx CRO offers expertise in study design, has affiliations with top clinical sites, provides regulatory consulting for EU standards, and ensures end-to-end study monitoring to maintain the quality and integrity of data.
Is MDx CRO suitable for both startups and established manufacturers?
Absolutely! Whether you’re a startup entering the IVD market or a seasoned manufacturer, MDx CRO’s tailored solutions cater to the unique needs of every client.
How does MDx CRO ensure compliance with the IVDR and ISO 20916?
MDx CRO boasts a regulatory consulting wing with deep knowledge of IVDR and ISO 20916, ensuring manufacturers receive accurate guidance and assistance throughout their IVD device’s journey to the market. Our team of former Notified Body experts on board help design studies that meet CE mark expectations
What advantages does MDx CRO offer in terms of site selection for IVD studies?
With its extensive experience and industry connections, MDx CRO has built relationships with leading clinical sites for a variety of technologies and clinical applications, ensuring timely and efficient study initiation and execution.
How does partnering with MDx CRO impact the success rate of IVD devices in the EU market?
With MDx CRO’s comprehensive services, from design to monitoring and regulatory guidance, manufacturers enhance their chances of a successful and compliant IVD product launch in the EU.
Where can I learn more about MDx CRO’s success stories or case studies?
It’s best to reach out to MDx CRO directly or visit our website for detailed testimonials, case studies, and more insights into our work.
Written by:
Carlos Galamba
CEO
Senior regulatory leader and former BSI IVDR reviewer with deep experience in CE marking high-risk IVDs, companion diagnostics, and IVDR implementation.
Companion Diagnostics IVD Consultancy within the EMA Framework: Comprehensive Guidance
August 8, 2023
The field of companion diagnostics IVD (CDx) represents a confluence of technological innovation, regulatory compliance, and patient care. As personalized medicine becomes an integral part of healthcare, the regulatory framework governing CDx, including the In Vitro Diagnostic Medical Devices Regulation (IVDR), has become more complex. This scenario calls for a specialized companion diagnostics consultancy. MDx CRO is at the forefront of this arena, offering expertise and guidance in the process for CDx consultation with the European Medicines Agency (EMA), Notified Body preparation and IVDR compliance within the European Union (EU).
Companion Diagnostics IVD and their Role
CDx are in vitro diagnostic (IVD) tests designed to provide information that is essential for the safe and effective use of a corresponding medicinal product. Their applications could include:
Identifying patients who are most likely to benefit from a particular therapeutic product.
Determining patients’ suitability for specific treatments.
Monitoring responses to ongoing treatments.
The Impact of IVDR on Companion Diagnostics
The IVDR sets out robust legal requirements for in vitro diagnostic medical devices, including CDx. Key aspects include:
Enhanced Patient Safety: Ensuring the quality and reliability of CDx IVDs.
Stricter Oversight: Increased scrutiny of the CDx development and approval process. Unlike the previous directive, CDx now require conformity assessment by a Notified Body, an independent organization designated to assess the compliance of medical devices and in-vitro diagnostics. In addition, CDx are also assessed by a medicines authority, most likely the EMA (European Medicines Agency), but a competent authority could also be involved .
Comprehensive Technical Documentation: Increased clinical evidence requirements are particularly notable in the IVDR. MDx CRO can help CDx manufacturers and their drug partners gather the necessary data to support their CDx application. This data may include clinical trial data (clinical performance data), analytical data, and safety data. Manufacturers must provide robust clinical evidence to demonstrate the performance, safety, and clinical utility of the CDx.
There are a number of other factors that can affect the approval process for CDx in the EU. These factors include:
The availability of data: Both the Notified Body and the EMA will need to have access to data from clinical trials that demonstrate the safety and effectiveness of the CDx.
The complexity of the CDx: The more complex the CDx, the more difficult it will be to assess its safety and effectiveness.
The novelty of the CDx: If the CDx involves new technologies or indications, the EMA and the Notified Body will need to take a more cautious approach to its approval. Different scenarios will play a role on the extent of scrutiny involved, including co-developed CDx scenarios, follow-on CDx, and CDx already on the market under the old IVD directive.
Understanding the EMA Companion Diagnostics Consultation Procedure
The consultation procedure is initiated by the notified body when it receives an application from a CDx manufacturer. The medicinal product involved could be a medicine already authorised for marketing in the EU or a medicine undergoing approval. Aligning drug and diagnostic development processes can help to ensure that the results of the clinical trials are accurate and reliable, and that the medicine is safe and effective when used with the CDx.
Aligning timelines in the drug and diagnostic (CDx) development process can help to ensure that the clinical trials for the medicine are conducted in a way that is consistent with the intended use of the CDx.
Upon application for a CDx IVD approval, the notified body will submit a letter of intent to the EMA, along with a technical dossier that describes the CDx and the medicinal product.
The EMA will then appoint a rapporteur, who will be responsible for reviewing the technical dossier and issuing a scientific opinion on the suitability of the CDx for use with the medicinal product. The rapporteur will also consider the views of any other interested parties, such as the applicant for the medicinal product, the manufacturer of the CDx, and patient groups.
The EMA will provide its scientific opinion on the CDx aspects that relate to the medicine to the notified body. The notified body will then use the EMA’s opinion to make a decision on whether to grant the CE mark to the CDx, in accordance with the regulatory requirements of the in vitro diagnostics regulation (EU IVDR).
EMA procedure timetables play a major role in the success of the consultation and turn around times for responses can be extremely short. Manufacturers should factor this in as they plan for their CDx submissions. There is the possibility to request a pre-submission meeting which will include representatives from Notified Bodies, EMA and could also include the drug manufacturer – this is used strictly to align on procedural and timing considerations (it is not used to provide feedback on study design or the content of the technical documentation).
One of the key documents used in the consultation and submitted by the notified body to the EMA is the SSP (Summary of Safety and Performance). The EMA expects manufacturers to use the SSP template provided in MDCG 2022-9. A lot more detail is expected in the SSP when compared to the information provided in the IFU. For example, detail on concordance studies is needed, particularly for co-developed CDx when different versions of a diagnostic have been used throughout the clinical development program.
MDx CRO: Your Partner in Companion Diagnostics Consultancy
Our companion diagnostics consultancy services encompass every stage of development, approval, and post-market surveillance:
Guidance on IVDR Requirements: In-depth support in understanding and meeting the specific demands of IVDR as they relate to CDx. MDx CRO can help a diagnostics company identify the specific requirements that apply to its CDx. For example, the requirements for a CDx that is intended to assess a patient’s suitability for treatment may be different from the requirements for a CDx that is intended to be used to monitor a patient’s response to treatment.
Preparation for Notified Body Assessment: Tailored strategies for successful assessment of a CDx under the IVDR: Assistance with compiling and submitting the necessary technical documentation and quality related documents.
Providing training to the manufacturer’s staff: MDx CRO can provide training to the manufacturer’s staff on the EMA’s requirements for CDx, as well as the notified body’s assessment process and expectations. This training will help to ensure that the manufacturer’s staff are prepared to answer any notified body questions and increase chances of success.
Stakeholder Communication: Facilitating communication with all relevant parties.
Global Perspective: Navigating international considerations for CDx in multi-country studies.
Post-Market Support: Focused on maintaining the highest standards through ongoing compliance monitoring with IVDR and other regulatory requirements. This includes implementing strong post-market surveillance processes and Post-Market Performance Follow-up (PMPF) evaluations, monitoring the CDx’s performance in real-world clinical settings, tracking and analyzing adverse events related to CDx usage, and conducting ongoing studies to evaluate the long-term impact and effectiveness of the CDx.
Why MDx CRO for Companion Diagnostics IVD Consultancy?
Expertise: Our in-depth knowledge of CDx, IVDR, and EU regulations offers unparalleled support.
Collaboration: Working closely with clients, we tailor our approach to meet specific needs.
Efficiency: Our insights and guidance save valuable time and resources, simplifying complex regulatory pathways.
Commitment: Our dedication to excellence, patient safety, and innovation sets us apart.
Navigating the multifaceted world of companion diagnostics in the EU, with the added complexity of IVDR, requires a dedicated and skilled partner. MDx CRO stands ready to be your guide in this critical journey, ensuring alignment with all regulatory standards. Reach out to explore how our companion diagnostics consultancy can be the key to unlocking your CDx potential in the EU’s dynamic regulatory environment.
FAQs
Q: What is a co-developed Companion Diagnostics in the context of EMA consultation?
A: A co-developed CDx is a device developed alongside a medicinal product for either initial authorization or a change of indication. This can include development during a pivotal clinical trial or a bridging study, with sufficient documentation to ensure performance alignment.
Q: How does a follow-on CDx differ from a co-developed CDx?
A: A follow-on CDx seeks the same indication as the original CDx but is not developed in parallel with the medicinal product. The follow-on CDx targets the same biomarker but may not be based on the same technology. It should be highly comparable to the original in performance, safety, and effectiveness.
Q: What documentation is required for a follow-on CDx?
A: Sufficient documentation must be provided for a follow-on CDx to prove that its analytical performance is comparable to the original CDx and that there’s no impact on clinical performance incompatible with the safe and effective use of the medicinal product.
Q: How are devices transitioning from IVDD to IVDR handled?
A: Devices initially marketed under Directive 98/79/EC (IVDD) that transition to IVDR fall under the co-developed or follow-on scenarios, depending on how they were initially developed.
Q: Is it possible to proceed with a single CDx consultation procedure for multiple authorized medicinal products and indications?
A: Yes, if a device’s intended purpose includes several authorized medicinal products and indications, it’s recommended to proceed with one single CDx consultation procedure. All concerned medicinal products should be listed in the intention to submit a letter by the Notified Body and in the application form.
Written by:
Carlos Galamba
CEO
Senior regulatory leader and former BSI IVDR reviewer with deep experience in CE marking high-risk IVDs, companion diagnostics, and IVDR implementation.
IVD Software Development: How to Bring IVD Software to Market in 8 Steps
April 4, 2023
The healthcare industry is undergoing a rapid transformation spurred by the advent of advanced medical diagnostic technology. IVD software development is a critical component of this revolution as it allows for testing, analysis, reporting, and communication without needing a physical laboratory or a visit to a doctor’s office.
Bringing IVD software development to the market can benefit patients and healthcare providers who can deliver quality care faster with fewer resources.
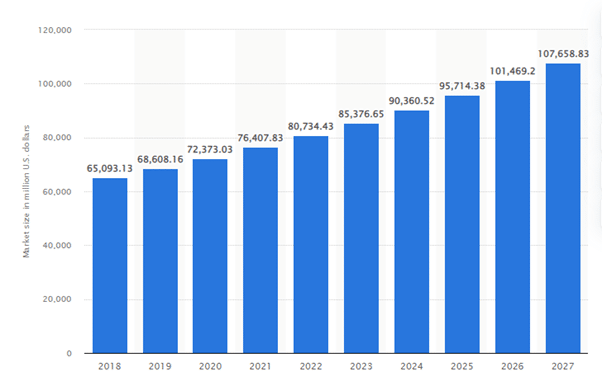
Projected size of the IVD market worldwide from 2018 to 2027 (in million U.S. dollars)
The above graph shows the In-Vitro Diagnostics (IVD) market globally was estimated at 72.4 billion U.S. dollars in 2020, with a projected growth of 108 billion U.S. Dollars by 2027, showing its increased relevance in the healthcare industry today.
1. Conduct Market Research
Before starting the development process, organizations must conduct market research and understand their target market, user needs, and potential competitors.
Market research should also help determine regulatory requirements that organizations must comply with and any current trends within the industry or technology space.
Below are some key points to consider when conducting market research for IVD software development:
Identify regulatory requirements: Identifying the regulatory requirements for your software is essential for bringing your product to market. They may vary depending on your target market, such as the FDA in the US or the European Union’s CE marking requirements (IVDR 746/2017).
Determine your target market: Identify the segments of the healthcare industry that your IVD software will serve. Consider factors such as geography, type of healthcare organization, and specialty areas.
Identify your competitors: Research the IVD software market to identify your competitors and their products. Analyze their strengths, weaknesses, pricing, and marketing strategies.
Understand your customers: Conduct surveys, interviews, and focus groups with healthcare professionals to understand their needs, preferences, and pain points. Use this information to tailor your IVD software to meet their specific needs.
Analyze market trends: Stay up-to-date on the latest trends and developments in the IVD software market. Monitor industry publications, attend conferences, and follow industry experts and thought leaders on social media.
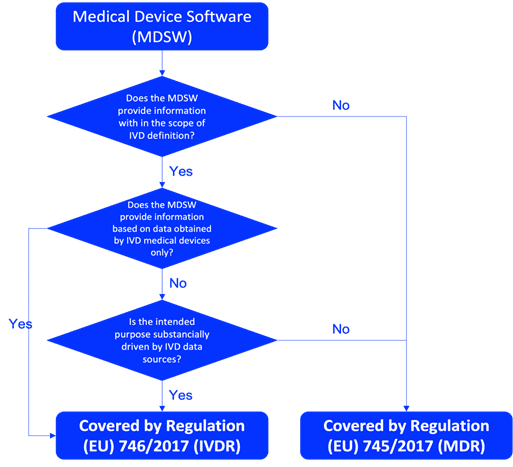
Understanding the classification of your IVD software is crucial before starting the planning and design process. Determine whether the IVD software is a standalone IVD medical device or a component of a larger system. To be qualified as an IVD medical device software in the EU, the product must fulfil the definition of IVD according to Article 2(2) of Regulation IVDR746/2017, as described in the Figure 1. The correct classification of IVD software should be done based on the rules described in Annex VIII of the IVDR. Relevant guidance documents, such as MDCG 2019-11 are also essentials for the qualification and classification of IVD software.
Fig. 1 – MDCG 2019-11 flowchart on qualification of Medical Device Software (MDSW)
Familiarize yourself with relevant regulatory frameworks, guidances and standards such as ISO 13485, IEC 62304, but also specific guidance documents published by regulators, which provide specifications and guidelines for developing, validating, and maintaining IVD software.
3. Plan and Design the Software
The next crucial step of a successful IVD software development is design and planning.
A well-documented and robust planning process can help provide a more detailed roadmap for development.
During this phase, design reviews, testing, and verification will ensure that the final version meets user requirements.
It is essential to incorporate user feedback at every stage of the design process to develop an intuitive interface that works effectively according to their needs.
Obtaining feedback from customers and stakeholders offers the development team opportunities to recognize potential concerns and areas for enhancement.
Developers can devise software solutions that fulfill customer needs and address their grievances by integrating feedback.
The importance of accurate documentation should not be underestimated as it helps trace back issues later on in the lifecycle of the software.
The development team must consider scalability and flexibility during the initial planning and design stages when creating the software.
4. Develop and Test the Software
Developing and testing the software is crucial in creating a working prototype.
The development phase is necessary to ensure the accuracy of the design, coding, and algorithms used in creating the software solutions.
Testing and quality assurance also play an essential role in ensuring the products meet all requirements before launch.
It is essential for companies to thoroughly assess each component of their software as part of this process. This includes ensuring they meet performance objectives concerning speed, responsiveness, scalability, security, reliability, and ease of use for their users.
Quality assurance checks help identify bugs or errors to release a defect-free product that meets all standards from regulatory bodies such as FDA or CE Marking.
When deploying IVD systems, manufacturers need to consider if their applications can be flexible enough to support new technological advances; future-proofing their products becomes increasingly necessary where customers demand longevity across upgrades or iterations over time.
5. Prepare a Regulatory Submission
Preparing a regulatory submission package is critical in bringing IVD software development to market. This step involves compiling documents to demonstrate that the software meets regulatory requirements and is safe and effective.
Here are some critical considerations for preparing a regulatory submission:
Gather relevant documents for the package
Understand regulations, standards, and risk classification of IVD software and the manufacturing role. Key documents to include are the device description, technical documentation, risk management file (ISO 14971), software lifecycle documentation (IEC 62304), and quality management system documentation (ISO 13485). Risk management must be applied and monitored during the IVD software development life cycle.
Prepare a performance evaluation report (PER)
This will require a comprehensive analysis of scientific evidence showing that the product meets user needs safely and effectively. IVD software performance evaluation should be prepared in accordance with relevant guidance documents, such as MDCG 2020-1 for the EU. Other guidance such as MedTech Europe Clinical Evidence Requirements for IVD can also be a good source of additional information.
Clinical performance studies are aimed at providing evidence of the safety and effectiveness of a product’s intended purpose to ensure that it’s able to diagnose, monitor and predict diseases and conditions accurately.
As described in MDCG 2020-1“Validation of the clinical performance should be considered at each change of the software to a new release. If no validation is performed, a justification should be stated in the technical documentation. With a validation of clinical performance, it is demonstrated that users can achieve clinically relevant outputs through predictable and reliable use of the MDSW”.
Adherence to relevant standards and guidelines, such as ISO 20916 (Clinical Performance Studies for In vitro diagnostics) and Good Clinical Practice (CGP), are crucial for the successful execution of clinical studies.
Ensure data accuracy
Ensure that that any data collected from testing is presented accurately to prove safety and efficacy before submitting your application. This includes validation and verification data, performance evaluations, and, if applicable, results from clinical studies. Carefully review all information for accuracy and completeness before submitting it.
6. Obtain Regulatory Approval
You need to obtain regulatory approval to bring IVD software development to market. Successful approval enables you to market and sell your software in compliance with local laws and regulations.
Familiarize yourself with the applicable regulatory frameworks and guidelines for your software. These may include ISO 13485 (quality management systems), IEC 62304 (medical device software lifecycle processes), ISO 14971 (risk management), and MDCG guidance documents such as MDCG 2019-11 (qualification and classification of software in the medical device regulations) and MDCG 2020-1 (guidance on performance evaluation for IVD software under the IVDR. You should also consider guidelines from other jurisdictions depending on your market strategy. The FDA for example has issued guidance for software as a medical device (SAMD).
Develop a comprehensive application package that should include all the necessary documents, data, and tests required for review. Ensure your package addresses regulatory requirements, such as conformity assessment procedures, clinical evidence, and post-market surveillance.
Submit the application package to the relevant regulatory authorities for review, such as the FDA in the US or notified bodies in the EU under the In Vitro Diagnostic Regulation (IVDR) 2017/746.
Be prepared to respond quickly and accurately to any feedback or additional information requests from regulatory agencies during the review process.
Make recommended changes swiftly as part of your submission to obtain approval from agencies successfully.
Once you receive approval from regulatory agencies, you can move forward with marketing and selling your software according to local laws & regulations, ensuring ongoing compliance with any post-market requirements.
7. Developing Marketing and Sales Strategies
Creating a successful marketing and sales strategy is essential for bringing IVD software to the market, it allows for faster positioning and gaining a competitive advantage. Make sure to develop a strong brand identity with messaging that resonates with your audience.
In addition, researching customer needs and understanding key industry trends can create a more targeted approach when it comes to the marketing of IVD software solutions, increasing your likelihood for success.
Make sure to use multiple channels such as paid advertising, email campaigns, social media and webinars to reach out to potential customers from diverse segments.
And last but not least, creating effective communication strategies to engage with customers throughout the sales cycle will also be key to promoting IVD products successfully.
8. Launch and Support the Software
Launching and supporting software is a crucial element to its success. The product can be improved over time by providing regular updates and customer service, and users can get the best experience.
Here are some points to consider when launching your IVD software development:
Create a comprehensive support plan that puts customer needs first. Ensure you have an efficient process for handling inquiries and technical issues as they arise.
Ensure that all necessary software updates are completed on schedule, so users don’t experience any delay in accessing the product’s full features or bug fixes.
Assess the regulatory impact of changes and bug fixes, changes to your IVD software may or may not be significant. Consider whether changes impact your regulatory approvals. MDCG 2022-6 provides additional guidance on changes to design and intended purpose in the context of the new transition timelines for IVDs in Europe.
Set up user feedback forms or surveys so customers can share their thoughts on the product’s performance and what improvements they want to see. This will help drive further development of the software over time.
Offer ongoing training opportunities for new features, so users feel confident using them once released. This will also ensure that customers know how to use their investment in your IVD software development solution fully.
Revolutionizing Healthcare With IVD Software Development
In vitro diagnostic (IVD) software development has transformed the healthcare industry by providing cost-effective testing, analysis, reporting, and communication solutions without physical laboratory equipment.
Studies indicate that healthcare providers highly prioritize in vitro diagnostic (IVD) procedures, and their optimization has the potential to enhance patient outcomes. Therefore, the development of IVD software is crucial in facilitating quicker and more accurate diagnostic results, ultimately leading to the optimization of healthcare practices.
If you need a partner in IVD software development for your business, MDx CRO is an IVD consultancy that provides end-to-end solutions to accompany you at each step of the process. Our team of highly experienced CRO strategists has extensive expertise in bringing innovative medical devices and IVD technologies to market. Request your expert consultation today.
FAQs
What are the key considerations when designing IVD software?
There are several key considerations that companies should keep in mind when designing IVD software: user requirements, regulatory requirements depending on the target geographic location, data accuracy and effective data management, the software’s ability to integrate with other systems, as well as performance and usability.
What are the regulatory requirements for IVD software development in Europe?
The regulatory requirements for IVD software development in Europe are determined by the In Vitro Diagnostic Regulation (IVDR), which became applicable on May 26, 2022. They include, but are not limited to design and development, risk management, validation and verification, as well as compliance with GDPR.
What are the most common challenges in IVD software development?
The most common challenges in IVD software development include regulatory compliance (which can be complex and challenging to navigate through), ensuring integration compatibility with other systems, effective data management, and great user experience, among others.
How do you ensure the quality and reliability of IVD software?
To ensure the quality and reliability of IVD software, it’s important that companies follow all regulatory guidelines applicable to their geographical location, and use a quality management system to ensure that the development process is well-documented. Conducting testing, validation and verification processes is another essential element of software development for in vitro diagnostics.
What to Consider When Developing an IVD Clinical Performance Study for IVDR Compliance
March 9, 2023
In vitro diagnostic (IVD) devices are essential in healthcare as they provide accurate and reliable diagnostic information to healthcare providers. The development of an IVD device involves several stages, including research and development, design and prototyping, verification and validation, regulatory approval, and commercialization.
One of the critical steps in IVD development is the conduct of an IVDR clinical performance study to generate reliable and meaningful data to support regulatory approval and the device’s commercial success. In Europe, the in-vitro diagnostic regulation (EU IVDR) is now in force and all new products to market must meet very strict requirements of clinical performance.
The role of ISO 20916 in IVDR clinical performance studies
The design and execution of an IVD clinical performance study are critical to its success, and several factors must be considered to ensure that the study generates reliable and meaningful data. The International Organization for Standardization (ISO) has developed ISO 20916, a standard that provides guidance on the design and conduct of clinical studies for IVD medical devices. The standard is intended to help manufacturers, regulators, and other stakeholders ensure that IVD clinical performance studies are designed and conducted in a consistent and scientifically rigorous manner.
ISO 20916 covers several important aspects, including study design, sample size determination, selection of appropriate endpoints, statistical analysis, and reporting of study results. The standard emphasizes the importance of designing studies that are appropriate for the intended use of the IVD device and that incorporate good clinical practice (GCP) principles.
The plan should specify the study objectives, inclusion and exclusion criteria for study participants, study endpoints, and statistical analysis plan, amongst many other requirements. It should also include procedures for data management and quality control to ensure the accuracy and reliability of the data collected.
Another important aspect of ISO 20916 is the requirement to ensure the safety and well-being of study participants. The standard emphasizes the importance of obtaining informed consent from study participants and protecting their privacy and confidentiality. The standard also requires that studies be conducted in compliance with ethical principles and regulatory requirements.
Alignment with EU IVDR
In addition to ISO 20916, the implementation of the EU IVDR has increased the importance of conducting IVD clinical performance studies as they are required for regulatory compliance. The IVDR replaced the previous In Vitro Diagnostic Directive (IVDD) and introduced more stringent requirements for IVD devices, including clinical evidence requirements. IVD manufacturers are now required to demonstrate clinical evidence that supports a device’s intended purpose and its’ safety and performance. This is particularly important, because insufficient clinical evidence could ultimately lead to a product refusal at the Notified Body resulting in additional costs and delays to bringing product to market.
Amongst many requirements, an IVDR clinical performance study is designed and conducted in compliance with GCP principles. ISO 20916 has additional requirements, and both the regulation and the standard should be considered by all diagnostic manufacturers when developing clinical performance study plans or protocols.
How can MDx CRO help?
MDx is a Medical Device & IVD Contract Research Organization (CRO) that can help IVD device manufacturers with their clinical performance studies by providing a range of services, including:
study design
site selection
patient recruitment
study monitoring
data management
statistical analysis
MDx has extensive experience in conducting clinical performance studies for IVD devices and a deep understanding of the regulatory requirements for these studies. Our team of professionals is well-trained and experienced in managing all aspects of the study, from protocol development to study execution and data analysis. We work closely with our clients to ensure that their studies are designed and conducted in compliance with applicable regulations and guidelines and that they generate reliable and meaningful data.
Conclusion
Conducting an IVD clinical performance study is a critical step in the development and commercialization of an IVD device. By following best practices, working with experienced professionals, and selecting the right CRO, IVD device manufacturers can generate reliable and meaningful data that can support regulatory approval and the device’s commercial success, ultimately benefiting patients and healthcare providers. Adherence to the IVDR and the ISO 20916 standard can help ensure that the data generated is acceptable for regulatory submission and meets the safety and performance requirements for IVDs.
Gain access to MDx’s Precision Medicine Services Pack—detailing our capabilities in clinical research, regulatory strategy, and companion diagnostics. Learn how we support pharma, CDx, and advanced therapy developers across the clinical and regulatory lifecycle.