EU AI Act and Medical Devices: What SaMD Developers Need to Know (2026)
March 9, 2026
The EU AI Act (Regulation (EU) 2024/1689) entered into force on 1 August 2024 and is being phased in progressively through 2026 and beyond. For companies developing AI-powered Software as a Medical Device (SaMD), it introduces a second, overlapping regulatory obligation that runs alongside, and interacts with, the existing requirements of EU MDR and IVDR.
This is not a distant compliance horizon. The provisions most relevant to medical device AI became applicable from August 2026. Companies that have not yet assessed their AI systems against the AI Act risk gaps in their technical documentation and conformity processes at exactly the moment Notified Bodies are beginning to incorporate AI Act considerations into their assessments.
This guide explains what the AI Act requires from SaMD developers, how it interacts with MDR and IVDR, and what practical steps manufacturers should be taking now.
The AI Act applies to AI systems placed on the market or put into service in the EU. An AI system is defined as a machine-based system that, given explicit or implicit objectives, infers from inputs how to generate outputs such as predictions, content, recommendations, or decisions that can influence real or virtual environments.
This definition is intentionally broad. It covers:
Deep learning systems including convolutional neural networks used in medical imaging
Natural language processing tools used in clinical documentation or decision support
Bayesian classifiers and other probabilistic inference systems
It does not cover:
Traditional rule-based software with no learning or inference component
Software that executes fixed logic without adaptive behaviour
If your SaMD uses any form of machine learning or statistical inference to generate clinical outputs, the AI Act almost certainly applies.
2. High-Risk AI Classification for Medical Devices
The AI Act categorises AI systems by risk level. For medical device manufacturers, the critical category is high-risk AI.
Under Annex III of the AI Act, AI systems intended to be used as safety components of medical devices, or which are themselves medical devices regulated under MDR or IVDR, are automatically classified as high-risk AI.
This means: if your SaMD is a CE-marked medical device or IVD, or is a software component that performs a safety function within one, it is high-risk AI under the AI Act. There is no further classification analysis required, the medical device status determines it.
High-risk AI systems are subject to the full obligations of the AI Act, including:
Risk management system: an AI-specific risk management process, documented and integrated with the ISO 14971 risk management already required under MDR
Data and data governance: training, validation, and testing datasets must be relevant, representative, free of errors, and sufficiently complete; demographic and geographic representativeness must be documented
Technical documentation: a detailed record of the AI system’s design, development process, training methodology, validation approach, and performance characteristics
Transparency and instructions for use: users must be provided with clear information about the AI system’s capabilities, limitations, accuracy metrics, and circumstances under which human oversight is required
Human oversight: the system must be designed to allow human oversight and intervention; it must not undermine the ability of the operator or user to override, disregard, or reverse outputs
Accuracy, robustness, and cybersecurity: performance must be declared and validated; the system must be resilient to errors, faults, and adversarial manipulation
Conformity assessment: high-risk AI systems must undergo a conformity assessment before being placed on the market
3. How the AI Act Interacts with MDR and IVDR
This is where the compliance picture becomes complex, and where early planning pays off.
The AI Act does not replace MDR or IVDR. Both regulatory frameworks apply simultaneously to AI-powered SaMD. However, the EU has designed a streamlined pathway for medical devices that are already subject to Notified Body review under MDR or IVDR.
Under Article 11 and Annex II of the AI Act, AI systems that are regulated as medical devices benefit from a single technical documentation approach meaning the AI Act technical documentation requirements can be integrated into the existing MDR/IVDR technical file rather than creating a separate document set.
Similarly, for Class IIb and III medical devices (MDR) and Class C and D IVDs (IVDR) which are the most likely to contain high-risk AI the Notified Body involvement already required under MDR/IVDR can cover the AI Act conformity assessment. The Notified Body acts as the relevant conformity assessment body for both frameworks.
In practice this means:
What changes for AI-powered SaMD under the AI Act:
Technical documentation must now explicitly address AI-specific elements: training data governance, model validation across subgroups, bias assessment, explainability approach, and human oversight mechanisms
Post-market monitoring must include AI performance monitoring tracking model drift, accuracy degradation over time, and distribution shift in real-world data
Transparency obligations require new IFU content describing AI limitations and human oversight requirements
A fundamental rights impact assessment may be required for certain high-risk AI applications in healthcare
What does not change:
The MDR/IVDR conformity assessment route remains the primary pathway
The Notified Body relationship established for MDR/IVDR CE marking remains the relevant body
ISO 14971 risk management, IEC 62304 lifecycle management, and clinical evaluation requirements are unchanged AI Act risk management is additive, not a replacement
4. General Purpose AI (GPAI) Models in Medical Devices
A separate and increasingly relevant category is General Purpose AI (GPAI) large foundation models or multimodal AI systems that can be adapted or fine-tuned for specific applications.
If a SaMD developer is building on top of a GPAI model: for example, fine-tuning a large language model for clinical documentation, or adapting a vision foundation model for medical image analysis both the GPAI model provider and the SaMD developer have obligations under the AI Act.
GPAI model providers must publish technical documentation and comply with copyright and transparency requirements. SaMD developers who deploy or fine-tune GPAI models are responsible for ensuring the resulting system meets all high-risk AI obligations, including data governance, validation, and clinical performance claims. The validation methodology for fine-tuned GPAI models in medical contexts is an area where regulatory guidance is still developing, early engagement with your Notified Body is strongly recommended.
5. Key Timelines
August 2024: AI Act enters into force.
February 2025: Prohibitions on unacceptable-risk AI systems apply. Not directly relevant for medical SaMD, but important for any AI used in patient-facing administrative processes.
August 2025: GPAI model obligations apply. SaMD developers building on foundation models must assess their exposure now.
August 2026: High-risk AI obligations fully apply. This is the key deadline for medical device AI. From this date, new AI-powered SaMD placed on the EU market must comply with all high-risk AI requirements.
Post-2026: Notified Bodies designated under the AI Act will begin conducting AI Act-specific conformity assessments. The intersection with MDR/IVDR NB assessments will become a standard part of the conformity process.
6. What to Do Now: A Practical Checklist
Classify your AI systems. Identify every AI component in your SaMD portfolio and confirm whether it meets the EU’s definition of an AI system. For each, document the risk classification and the rationale.
Assess your technical documentation gaps. Review your existing MDR/IVDR technical files against the AI Act Annex IV requirements. Identify where AI-specific content, training data documentation, bias assessment, explainability approach, is missing or insufficient.
Review your data governance. The AI Act’s requirements for training data representativeness and bias documentation are more explicit than anything in MDR. If your training data governance is not documented at the level the AI Act requires, this is a gap that needs addressing before your next Notified Body audit.
Update your IFU and labelling. Transparency obligations mean users must be explicitly informed about AI limitations, performance metrics across relevant subgroups, and circumstances requiring human override. Most current SaMD IFUs are not written to this standard.
Engage your Notified Body. Ask your NB directly how they are approaching AI Act integration into MDR/IVDR assessments. Different NBs are at different stages of readiness, and early clarity on what they will expect prevents last-minute documentation gaps.
Build AI performance monitoring into your PMS. Post-market surveillance for AI-powered SaMD must now track model performance over time. If your PMS plan does not include AI-specific monitoring metrics, update it before August 2026.
Regulatory affairs specialist & CRA with expertise in EU MDR/IVDR, CE marking, Biological Evaluations (dental), and clinical investigations & technical documentation for MDs & IVDs.
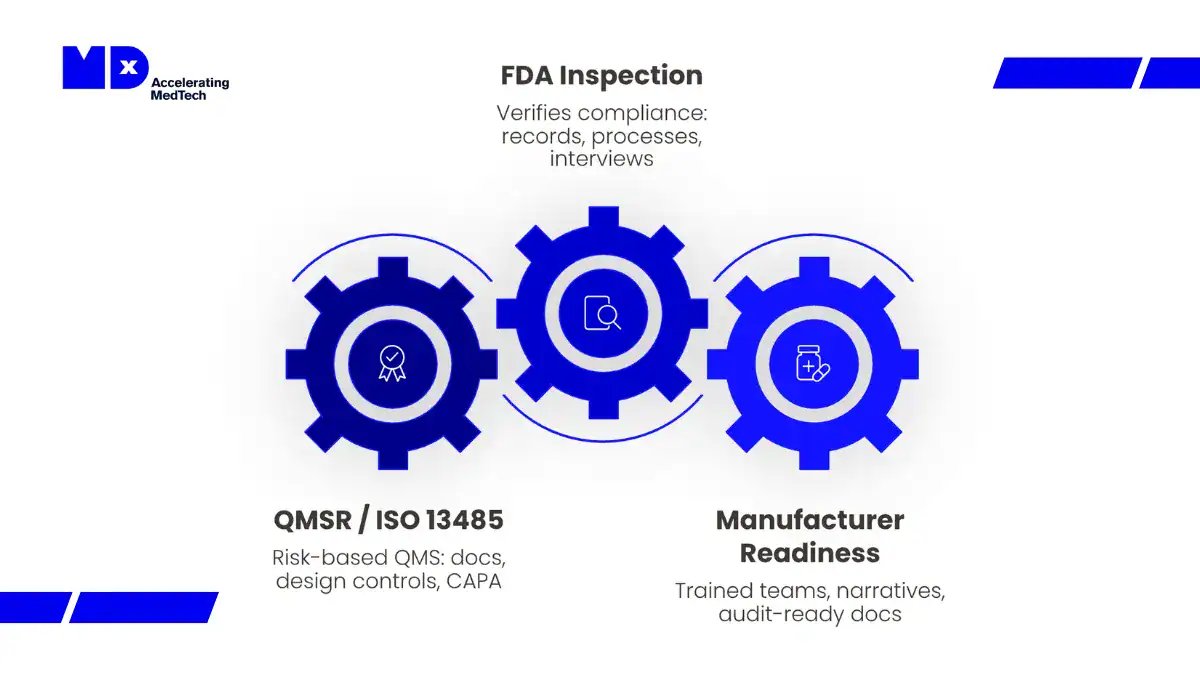
This article explains what the FDA QMSR changes in practical terms.
Specifically, it clarifies:
How the updated MDSAP Audit Approach aligns with QMSR
How FDA inspections now operate under CP 7382.850
Which documentation areas receive increased inspection attention
How manufacturers can structure a QMSR gap assessment
What inspection exposure points regulatory teams should proactively address
These changes are particularly relevant for manufacturers operating in complex regulatory environments, including companies developing FDA companion diagnostics. In these companies design controls, labeling, clinical evidence, and post-market monitoring must operate as an integrated lifecycle system.
Why the FDA QMSR Matters Now?
The FDA QMSR represents more than structural alignment with ISO 13485. It modernizes inspection methodology, expands documentation accessibility, and reinforces a lifecycle-based, risk-driven enforcement approach.
Under the former QSR model, inspections often focused on subsystem compliance using QSIT checklists. Under QMSR, investigators evaluate how quality processes function collectively across the total product lifecycle.
MDSAP Audit Approach 2026: How It Differs from FDA Inspections Under QMSR
MDSAP audits and FDA inspections are frequently discussed together. However, they serve fundamentally different regulatory functions.
An MDSAP audit is conducted by an FDA-recognized Auditing Organization. It provides a structured, scheduled, and standardized assessment covering ISO 13485 and the regulatory requirements of participating authorities, including the FDA. The audit focuses on conformity with harmonized quality management system requirements. Participation remains voluntary.
An FDA inspection, by contrast, is a statutory enforcement activity conducted directly by FDA investigators. Its objective is not certification, but the evaluation of compliance with U.S. legal requirements and the identification of potential violations, systemic weaknesses, or public health risks.
Under the FDA QMSR framework, this distinction remains critical.
In practical terms:
MDSAP is conformity-focused, scheduled, and audit-driven.
FDA inspections are compliance-driven, investigative, and enforceable.
Because of these structural differences:
MDSAP does not replace FDA’s legal authority to inspect.
FDA inspections are not limited by the MDSAPtask structure.
A successful MDSAP audit does not guarantee a favorable FDA inspection outcome.
While strong MDSAP performance may influence FDA surveillance planning, it does not eliminate the possibility of routine, risk-based, or for-cause inspections.
Common Industry Assumptions That Elevate Inspection Risk Under QMSR
Despite years of MDSAP implementation and increasing alignment with ISO 13485, several structural misunderstandings remain common within regulatory teams. Under the FDA QMSR framework, these assumptions may create unintended inspection exposure.
Among the most frequent are:
“MDSAP replaces FDA inspections.” MDSAP may inform FDA’s surveillance planning. However, it does not limit FDA’s statutory authority to conduct routine, risk-based, or for-cause inspections.
“If the MDSAP auditor did not identify it, FDA will not pursue it.” FDA investigators are not constrained by MDSAP audit depth, sampling methodology, or task sequencing. They may pursue any line of inquiry necessary to evaluate U.S. compliance.
“MDSAP and FDA inspections evaluate the same criteria.” MDSAP assesses conformity to harmonized quality system requirements. FDA inspections evaluate compliance with U.S. law and potential public health impact.
“ISO 13485 certification ensures FDA compliance.” Although ISO 13485 is incorporated by reference into QMSR, FDA-specific statutory and regulatory requirements remain fully enforceable.
“MDSAP covers all U.S.-specific expectations.” While the audit model maps U.S. requirements, FDA inspections may extend beyond mapped tasks and request additional evidence where risk or compliance concerns arise.
Collectively, these assumptions overlook a central element of the FDA QMSR transition.
QMSR vs QSR vs ISO 13485: What Actually Changed
Many organizations assume that QMSR simply “equals ISO 13485.” However, that interpretation is incomplete.
Although ISO 13485:2016 is incorporated by reference into U.S. law, FDA-specific statutory and regulatory requirements remain fully enforceable. Therefore, obligations related to UDI, Medical Device Reporting (MDR), device listing, and labeling controls continue to apply.
As a result, companies that focus solely on ISO 13485 conformity may overlook additional documentation, traceability, and enforcement expectations that FDA investigators apply during inspections.
Comparison: QSR vs QMSR vs ISO 13485
Element
Former QSR (21 CFR 820)
FDA QMSR (2026)
ISO 13485:2016
Legal Status
U.S. regulation
U.S. regulation incorporating ISO 13485 by reference
International standard (voluntary unless required)
Inspection Model
QSIT-based
CP 7382.850 lifecycle & risk-based
Certification audit model
Internal Audit Access
Certain protections under §820.180(c)
Internal audit & management review records reviewable
Auditor access during certification
CAPA Focus
Procedural compliance
Demonstrated effectiveness & root cause verification
Effectiveness required but certification-oriented
Design Controls
Detailed QSR-specific structure
ISO 13485 clause 7.3 structure + FDA overlays
Clause 7.3 framework
FDA-Specific Requirements
Fully embedded
Still fully enforceable (UDI, MDR, labeling, etc.)
Not included
Key takeaway: ISO 13485 alignment does not eliminate FDA-specific compliance obligations. FDA retains enforcement authority under U.S. law.
FDA Compliance Program 7382.850: How Inspections Work Under QMSR
As of February 2, 2026, FDA retired QSIT and implemented Compliance Program 7382.850.
Inspections now organize around:
Six Quality Management System (QMS) Areas:
Change Control
Design & Development
Management Oversight
Outsourcing & Purchasing
Production & Service Provision
Measurement, Analysis & Improvement
Four Other Applicable FDA Requirements (OAFRs):
Tracking
Corrections & Removals
Medical Device Reporting (MDR)
Unique Device Identification (UDI)
Under this model, FDA evaluates how quality subsystems operate as an interconnected framework rather than as isolated elements. Inspectors assess whether risk information, design decisions, post-market data, and management oversight are aligned throughout the product lifecycle.
For manufacturers of companion diagnostics, this system-level evaluation is particularly significant, as design inputs, labeling claims, clinical performance data, and post-market monitoring directly influence one another.
What FDA Inspectors Scrutinize Most Under QMSR
Based on regulatory inspection support experience, three documentation areas now present heightened exposure.
1. Internal Audits, Supplier Audits, and Management Review Records
Under QMSR, FDA inspectors may review:
Internal audit reports
Supplier audit outcomes
Management review records
As our QA/RA Specialist Joana Martins notes from inspection support experience, investigators increasingly evaluate whether quality processes function effectively in practice, not merely whether procedures formally exist.
Records must clearly demonstrate:
Identified issues
Root cause analysis
Corrective actions
Follow-up and documented closure
Incomplete or draft audit records increase inspection risk.
2. Design Controls and Traceability (ISO 13485 Clause 7.3)
QMSR aligns with ISO 13485 clause 7.3. However, manufacturers must demonstrate full traceability across:
User needs
Design inputs
Design outputs
Verification and validation
Residual risks
Traceability weaknesses frequently arise between:
Risk management files
Labeling claims
UDI triggers
MDR criteria
Inspectors expect objective evidence that these elements remain consistently aligned across documentation and decision-making processes.
For companion diagnostics, this alignment is especially critical because intended use, biomarker claims, and clinical evidence directly impact regulatory risk classification.
3. CAPA and Effectiveness Verification
CAPA remains one of the most enforcement-sensitive areas under QMSR.
A recurring weakness observed during inspection preparation is the absence of documented effectiveness verification following corrective actions. Closing a CAPA administratively is insufficient. Investigators expect objective evidence demonstrating that actions eliminated root causes and prevented recurrence.
Documented effectiveness checks are not procedural formalities, they serve as evidence that the quality system operates as intended.
Inspection Risk Indicators Under FDA QMSR
The transition to the FDA QMSR has altered not only inspection structure but also inspection depth. Under Compliance Program 7382.850, FDA investigators apply a lifecycle and risk-based model that prioritizes system effectiveness, data integrity, and management oversight.
For this reason, manufacturers should not wait until an inspection is scheduled to evaluate potential vulnerabilities. Identifying structural weaknesses in advance is critical because inspection findings under the QMSR framework increasingly derive from systemic inconsistencies rather than isolated documentation gaps.
Proactive identification of inspection risk indicators allows organizations to:
Reduce the likelihood of Form 483 observations
Prevent escalation to warning letters or enforcement action
Shorten remediation timelines
Demonstrate mature quality governance
Below are recurring inspection risk indicators observed in practice under the evolving QMSR inspection model:
Examples of Documentation Weaknesses Observed During Inspections
Risk Area
Typical Vulnerability
CAPA
Repeated issues without documented effectiveness verification
Design Controls
Incomplete traceability between risk analysis and design inputs
Management Review
Minutes lacking documented decisions, metrics, or follow-up actions
Supplier Oversight
Absence of risk-based justification for audit scope
Post-Market Surveillance
Complaint trends not connected to CAPA or design updates
Documentation Areas Receiving Increased Inspection Attention Under QMSR
The transition to QMSR expands the practical scope of documentation that investigators may review.
Under the former QSR, certain internal records were less frequently examined due to inspection structure and interpretative practice. Under QMSR, those same records may serve as direct evidence of whether management oversight, supplier controls, and internal audit processes operate effectively.
Investigators assess whether:
Issues are identified systematically
Root causes are documented clearly
Decisions are traceable
Corrective actions are verified for effectiveness
Inconsistent documentation, incomplete audit closure, or lack of traceability between systems may now carry greater inspection consequences than under the previous model.
QMSR Gap Assessment Framework
ISO 13485 certification does not automatically confirm FDA QMSR compliance. A structured gap assessment helps identify regulatory overlays and inspection exposure points.
A practical QMSR gap assessment should include:
1. Clause Mapping
Map ISO 13485 clauses to QMSR references and confirm terminology alignment.
2. FDA-Specific Overlay Identification
Verify incorporation of:
UDI requirements
MDR reporting triggers
Labeling obligations
Device listing controls
3. Documentation Exposure Review
Assess:
Internal audit completeness
CAPA effectiveness evidence
Management review decision traceability
Supplier risk classification
4. Inspection Simulation
Conduct mock inspections aligned with CP 7382.850 to test system coherence.
Even when corrective actions remain in progress, documented identification and remediation planning demonstrate regulatory control and transparency.
What the QMSR Means for U.S. Manufacturers and FDA Inspections
The most important U.S.-specific change is the removal of references to the former FDA Quality System Regulation (21 CFR 820). These have been replaced with references aligned to the QMSR, under which ISO 13485:2016 is now incorporated by reference into U.S. law. This does not mean that all FDA-specific requirements disappear. U.S. statutory and regulatory obligations continue to apply where relevant.
The updated MDSAP Audit Approach also reflects several U.S. regulatory updates that have been in effect since March 2024:
Device listing updates: Manufacturers must confirm or update their device listing information annually between October 1 and December 31, or whenever a relevant change occurs (21 CFR 807).
Predetermined Change Control Plans (PCCPs): The audit model now clarifies how PCCPs are assessed, particularly for software-based and AI-enabled devices, aligning with FDA’s existing change control requirements under 21 CFR 807.81 and 21 CFR 814.39.
These requirements are not new, but the 2026 MDSAP update removes inconsistencies between what auditors assess and what FDA expects.
Beyond the U.S.-specific updates, there is also a broader change that affects all participating jurisdictions. The term “critical supplier” has been removed and replaced with more practical language referring to “suppliers that should be considered for audit as part of the MDSAP audit of the organization.” This better reflects ISO 13485 risk-based thinking and reduces ambiguity around supplier oversight across different regulatory systems.
Other international changes Under the FDA’s QMSR Framework
While this article focuses on the U.S. regulatory framework, the updated audit approach also incorporates important revisions from other participating regulatory authorities:
Australia (TGA): Alignment with the Procedure for Recalls, Product Alerts and Product Corrections (PRAC), which came into effect in March 2025.
Brazil (ANVISA): Updated references to RDC 830/2023 (IVDs) and RDC 751/2022 (medical devices).
These changes ensure the audit model reflects current regulatory frameworks across all participating jurisdictions.
Practical implications for medical device manufacturers
FDA’s new compliance program significantly raises expectations for how manufacturers demonstrate quality system effectiveness during inspections:
FDA inspectors are evaluating how quality processes work together in practice. Making a strong emphasis on risk management, data integrity, and decision-making across the total product lifecycle.
Previously “internal” records are now fair game. Internal audit reports, supplier audit outcomes, and management review records may be reviewed during inspections. These documents must clearly reflect issues identified, decisions made, and actions taken.
Risk management must be continuous and demonstrable. FDA expects risk to be actively monitored and linked to CAPA, design changes, supplier controls, and post-market surveillance, and not treated as a static or one-time exercise.
Post-market data is a primary inspection focus. Complaint trends, medical device reporting, recalls, UDI, and tracking data are increasingly used to assess whether the quality system is effective and responsive to real-world performance.
Inspection scope may be driven by data. FDA may use pre-inspection data reviews or remote assessments to target areas of concern, increasing scrutiny where trends or inconsistencies are identified.
To summarize, manufacturers should ensure their quality systems tell a coherent, data-supported story and demonstrate not just compliance, but control and effectiveness.
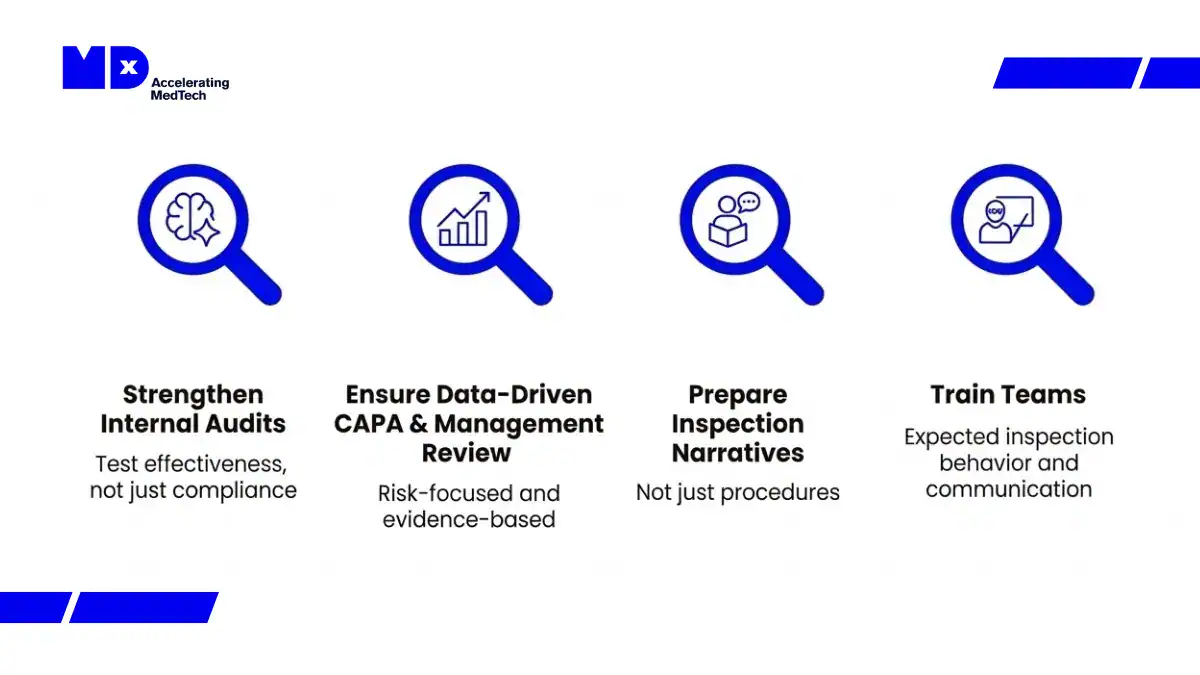
What medical device manufacturers should do before their next audit or inspection
Manufacturers should:
Strengthen internal audits to test effectiveness, not just compliance
Ensure management review and CAPA are data-driven and risk-focused
Prepare clear inspection narratives, not just procedures
Train teams on inspection behavior and communication
By strengthening these foundations, manufacturers can approach their next audit or inspection with clarity, confidence, and control.
Key takeaways for companies targeting the US market
MDSAP remains valuable, but it is no longer sufficient on its own
FDA inspections are becoming more structured, consistent, and data-driven
Early alignment with QMSR expectactions reduces inspection risk, delays and remediation costs
Strategies for Modern and effective compliance.
How MDx Supports FDA QMSR Readiness: Expert Insight
Transitioning from QSR to FDA QMSR requires more than updating terminology. It demands structural alignment, inspection-oriented preparation.
Based on field experience supporting manufacturers through inspection preparation and regulatory alignment projects, Joana Martins, QA/RA Specialist at MDx, emphasizes that the most frequent vulnerabilities do not stem from missing procedures, but from insufficiently demonstrated system effectiveness.
According to Joana’s inspection readiness experience, organizations often underestimate three exposure points during FDA inspection preparation:
The depth of documentation review now permitted under QMSR
The need for traceability between risk management, design controls, and post-market data
The importance of documented effectiveness verification within CAPA systems
To address these exposure points, MDx supports medical device manufacturers through:
Independent QMSR-aligned readiness assessments focused on inspection exposure
Structured QMSR gap analysis incorporating FDA-specific regulatory overlays
Mock FDA inspections aligned with Compliance Program 7382.850
Strategic support for companies developing FDA companion diagnostics, where design traceability, labeling controls, and lifecycle data integration require heightened regulatory coherence
Rather than approaching FDA QMSR as a documentation update, MDx works with organizations to ensure their quality systems demonstrate operational integrity, risk-based decision-making, and inspection resilience.
Organizations preparing for FDA inspection or evaluating their QMSR alignment can benefit from early, structured assessment. Proactive evaluation reduces remediation timelines, minimizes inspection disruption, and strengthens regulatory confidence.
Frequently Asked Questions About FDA QMSR, MDSAP, and Inspections
What is the main difference between QSR and QMSR?
The main difference is structural alignment. Under QSR, FDA requirements were written directly into 21 CFR Part 820. Under QMSR, the FDA incorporates ISO 13485:2016 by reference into U.S. law while keeping FDA-specific obligations in force. In short, QMSR harmonizes structure with ISO 13485. However, it does not reduce FDA enforcement authority or eliminate U.S.-specific requirements such as MDR, UDI, or device listing.
Does ISO 13485 certification guarantee FDA compliance under QMSR?
No, it does not. Although ISO 13485 forms the backbone of QMSR, FDA-specific statutory requirements still apply. Manufacturers must comply with MDR, UDI, corrections and removals, and other U.S. obligations. Based on regulatory experience, companies often assume ISO certification closes all gaps. In practice, a targeted QMSR gap assessment is necessary to confirm full FDA alignment.
What replaced QSIT in FDA inspections?
FDA replaced QSIT with Compliance Program 7382.850, effective February 2, 2026. This new program aligns inspections with the QMSR framework. Instead of subsystem checklists, FDA now organizes inspections around six QMS areas and four Other Applicable FDA Requirements (OAFRs). As a result, inspections follow a more integrated, risk-based, lifecycle-focused approach.
Can FDA inspect internal audit reports under QMSR?
Yes. Under QMSR, FDA investigators may review internal audit reports, supplier audits, and management review records. In practice, inspectors now verify whether issues were identified, documented, and effectively closed. They no longer focus only on whether procedures exist, they assess whether the system works as intended. Incomplete or unverified audit actions may increase inspection risk.
What records are now receiving greater scrutiny during FDA inspections?
FDA now places greater scrutiny on: – Internal and supplier audit reports – Management review documentation – Design control traceability records – CAPA procedures and effectiveness checks From inspection experience, CAPA effectiveness verification is a frequent weak point. Companies often implement corrective actions but fail to document objective evidence that the action resolved the root cause. Under QMSR, effectiveness matters as much as documentation.
Why are companies that passed MDSAP still receiving FDA 483 observations?
Because MDSAP and FDA inspections serve different purposes. MDSAP evaluates conformity. FDA inspections assess legal compliance and public health risk. FDA investigators are not bound by MDSAP sampling methods or audit scope. If inspectors identify ineffective CAPA, weak traceability, or gaps between procedures and actual practice, they may issue Form 483 observations, even after a successful MDSAP audit.
How should manufacturers prepare for FDA inspections under QMSR?
Start early. Preparation often takes longer than expected. Then conduct a structured QMSR gap assessment. ISO 13485 compliance alone does not confirm full FDA alignment. Finally, train teams on Compliance Program 7382.850. Mock interviews and inspection simulations help identify weaknesses. Even documented remediation in progress demonstrates system control and reduces inspection risk.
When does FDA QMSR enforcement begin?
FDA QMSR enforcement began on February 2, 2026, when the new Quality Management System Regulation officially replaced the former Quality System Regulation (21 CFR Part 820). From that date, FDA inspections operate under Compliance Program 7382.850.
What are the FDA QMSR and ISO 13485 harmonization requirements for 2026?
Under the 2026 QMSR, ISO 13485:2016 is incorporated by reference into U.S. law. This means manufacturers must meet ISO 13485 requirements as part of FDA compliance. However, harmonization is not complete equivalence, FDA-specific obligations such as UDI, MDR reporting, device listing, and labeling controls remain fully enforceable and are not covered by ISO 13485 alone. See the QSR vs QMSR vs ISO 13485 comparison table above for a detailed breakdown.
Written by:
Joana Martins
QARA Specialist
QA/RA Specialist supporting teams in clinical evaluations and regulatory compliance with ISO 13485, MDR, and international medical device requirements (FDA, Health Canada, ANVISA).
The evolving landscape of Companion Diagnostics (CDx) introduces complexities in regulatory and certification processes. Engaging in IVDR Companion Diagnostic Consulting is essential to ensure a streamlined and compliant journey.
Deciphering Regulatory Nuances: US vs. EU
Historically, CDx devices in the EU were self-certified under the IVDD. A CDx manufacturer may have had experience with the FDA but the regulatory process in the EU is only now emerging.
The EU IVDR defines a CDx as a device which is essential for the safe and effective use of a corresponding medicinal product to identify, before and/or during treatment:
Patients who are most likely to benefit from the corresponding medicinal product
Patients likely to be at increased risk of serious adverse reaction as a result of treatment with a corresponding medicinal product
The FDA’s definition is similar but extends to devices used for “monitoring treatment responses with a particular therapeutic product”. Unlike in the US such devices are not considered companion diagnostics in the EU. Furthermore, the FDA acknowledges a category of devices termed complementary diagnostics. These diagnostics are characterized as tests that pinpoint a group of patients, identified by specific biomarkers, who respond well to a drug. While they assist in evaluating the risk-benefit ratio for individual patients, they aren’t mandatory for drug administration. Within the IVDR framework, complementary diagnostics aren’t explicitly detailed, nor do they have specific prerequisites for CE certification
These nuances are key for any CDx regulatory strategy and for the planning of CDx clinical trials. Aspecialized IVDR CDx consulting company like MDx CRO can help diagnostic companies and their pharma partners navigate global differences and ensure CDx regulatory compliance.
The EMA Consultation Process
EMA’s guidance stands as a pivotal component in IVDR Companion Diagnostic Consulting. The EMA CDx Assessment Report Template, publicly available, provides a comprehensive blueprint. It is a great source of information for the expectations in CDx submission content, particularly useful for when drafting SSPs and IFUs.
Optional, but highly recommended, pre-submission meeting.
Application submission.
Interactive Q&A phases.
EMA’s final verdict.
Crafting of SSP & IFU with Detail
For successful IVDR CDx certification, the SSP and IFU documents should be meticulously detailed as they are the 2 key documents used during the EMA consultation process.
Diagnostic manufacturers should ensure they include:
Emphasis on scientific validity of the biomarker
Comprehensive detail on performance evaluation, study design descriptions, encompassing both analytical and clinical performance.
Insight into clinical data, detail on device modifications during or after the clinical performance study, and associated impacts, rationale for cut-off point selection and more.
A deep dive into the risk-benefit analysis is pivotal, concentrating on major residual risks and device limitations.
Time Considerations for IVDR CDx Certification
The certification process for CDx under IVDR is extensive. From the initial 3-month EMA notification to the concluding recommendation, the timeline can span 8-18 months. Such extended durations underline the criticality of early preparations. Engaging early with a specialized CDx consulting company can help avoid surprises and streamline the CDx certification journey.
The expertise offered by the notified body can significantly enrich IVDR Companion Diagnostic certification. Early engagements, prior to document submissions, can provide clarity, ensuring alignment with EMA requirements.
Selecting your IVDR CDx Consulting partner
MDx CRO has published a deep dive into the crucial factors to bear in mind when picking an IVD consultant.
In the dynamic realm of CDx, efficient navigation is paramount. If you’re seeking specialized insights into IVDR certification, explore our IVD services. At MDx CRO, our experts offers tailored IVDR Companion Diagnostic Consulting, ensuring optimal integration of CDx within the regulatory framework.
Contact our team today to discuss your CDx product needs!
Written by:
Carlos Galamba
CEO
Senior regulatory leader and former BSI IVDR reviewer with deep experience in CE marking high-risk IVDs, companion diagnostics, and IVDR implementation.
ISO 20916:2024 What You Need to Know for Clinical Performance Studies in 2026
February 2, 2026
Carlos Galamba
Planning a clinical performance study under IVDR in 2026 requires more than regulatory awareness, it requires strategic alignment with ISO 20916:2024.
This article breaks down what ISO 20916:2024 means in practice, how it interacts with IVDR, the key differences you must consider before designing your study, and how to position your project for regulatory success.
What changed with ISO 20916:2024 and how it affects your study strategy
ISO 20916:2024, Clinical performance studies using specimens from human subjects: Good study practice, was first introduced in 2019. In March 2024, it was published as EN ISO 20916:2024, marking a major step in ISO 20916 2024 IVDR harmonization IVD studies across Europe. This update aligns clinical performance study requirements directly with the IVDR.
In today’s rapidly evolving IVD landscape, safety and performance remain top priorities. ISO 20916 provides a solid framework to ensure clinical performance studies are planned, executed, recorded, and reported with scientific rigor. Its goal is simple: ensure IVD studies are ethical, reliable, and aligned with regulatory expectations.
This standard supports robust study design, promotes high‑quality data generation, and strengthens compliance for IVD manufacturers navigating the IVDR.
Planning a clinical performance study in 2026?
Request access to our expert session on preparing a GDD clinical performance study under ISO 20916:2024.
The March 2024 revision introduced a key milestone: Annex ZA, which formally harmonizes ISO 20916 with the IVDR. While the IVDR already referenced ISO 20916, the addition of Annex ZA creates a unified regulatory pathway for clinical performance studies.
Annex ZA bridges the remaining gaps between the standard and the regulation. As a result, manufacturers benefit from clearer expectations and a more predictable approval process.
EN ISO 20916:2024 was approved by CEN without modification, reinforcing its relevance for EU regulatory compliance. However, at the time of writing, official recognition of ISO 20916:2024 as an IVDR harmonized standard in the EU Official Journal is still pending.
Note: As of the date this article was written, the official recognition of ISO 20916:2024 as an IVDR harmonized standard for clinical performance studies in theEuropean Union’s Official Journal was awaiting confirmation.
How Annex ZA Connects ISO 20916:2024 with IVDR Requirements
1. Presumption of Conformity
Compliance with the ISO 20916 clauses listed in Table ZA.1 gives manufacturers a presumption of conformity with IVDR GSPRs. This presumption simplifies regulatory alignment across IVD clinical performance studies.
2. Definition Alignment
When definitions differ between ISO 20916 and the IVDR, Annex ZA prioritizes IVDR terminology. This ensures consistency across regulatory submissions.
3. Risk‑Management Updates
Annex ZA strengthens risk‑management expectations. It requires alignment with IVDR principles such as “reducing risks as far as possible.” It also notes that ISO 20916 does not include foreseeable misuse, while the IVDR does—requiring sponsors to bridge this gap.
4. Acceptable Risk Policies
Manufacturers must align acceptable risk decisions with specific GSPRs. Annex ZA also clarifies that while ISO 20916 excludes training as a risk‑reduction measure, the IVDR allows it.
ISO 20916 vs IVDR: A Practical Comparison for Study Design
Topic
ISO 20916
IVDR
Annex XIV studies
No specific terminology for surgically invasive sample taking
Recognizes surgically invasive sampling as Annex XIV study
Prescriptive; allows rationale for remote monitoring
Requires independent monitor
Informed consent
Highly detailed framework
Less detailed
Where ISO 20916 and IVDR Align
Despite some differences, the ISO and IVDR frameworks remain closely aligned in key areas:
Clinical performance parameters: Nearly identical, except ISO 20916 omits expected values for normal and affected populations.
Ethical considerations: ISO 20916 provides more detail, defining responsibilities for sponsors and investigators.
Bias mitigation: ISO 20916 offers explicit direction on preventing population, protocol, and reference‑method bias.
Site qualification: More detailed under ISO 20916, specifying resources, equipment validation, and QMS expectations.
CPSR content: ISO 20916 includes additional requirements, especially for interventional studies.
Comparator devices: The standard requires clear listing with commercial name, manufacturer, and catalog number.
Investigator’s Brochure: Both the IVDR and ISO 20916 are aligned, though ISO adds more detail on risk‑benefit documentation.
Who Should Apply EN ISO 20916:2024 in 2026?
Manufacturers of in vitro diagnostic medical devices
In vitro diagnostic clinics and laboratories
Test centres for in vitro diagnostic medical devices
Regulatory authorities
IVDR Notified Bodies
IVD Clinical research organizations (CROs)
Investigators and sponsors
Advantages of Applying EN ISO 20916 in IVD Performance Studies
Robust Results: It ensures high-quality, accurate, and reliable data generation, pivotal for safe healthcare decisions.
Ethical Standards: It upholds the rights, safety, dignity, and well-being of study subjects.
Study Planning and Conduct: It facilitates the meticulous planning and execution of IVD performance studies, ensuring regulatory and ethical compliance alongside scientific validity.
Compliance and Clarity: It provides a framework for compliance with IVDR, clarifying roles and responsibilities of all parties involved.
Risk Management: It emphasizes subject safety, especially regarding specimen collection risks, and ensures data integrity.
Implications for Sponsors and CROs Conducting IVD Performance Studies
The integration of ISO 20916 with the IVDR, highlighted by the inclusion of Annex ZA, significantly transforms IVD clinical performance studies and CRO operations. This crucial alignment demands a comprehensive revision in study design, execution, and reporting methodologies, highlighting the importance of compliance with the unified ISO 20916 and IVDR standards. It emphasizes the need for robust quality and risk management systems and ethically responsible study development.
This evolution signifies more than standard adherence; it represents a commitment to elevating IVD performance and efficacy in line with the highest EU regulatory standards. It requires IVD stakeholders, including CROs, sponsors and manufacturers, to deeply understand and agilely apply these standards, not only for compliance but to set new quality and safety benchmarks in diagnostics.
This commitment is fundamental to advancing patient care and public health, marking a significant step forward in regulatory compliance and industry excellence.
Since its foundation, MDx CRO has consistently used ISO 20916 as the benchmark for all our IVD clinical performance studies. The release of Annex ZA and its harmonization with IVDR reinforces our status as the leading CRO for IVD clinical performance studies.
Achieving Success in IVD Clinical Performance Studies
At MDx CRO, we navigate the complexities of IVDR and the latest ISO 20916 revision for in vitro diagnostic (IVD) studies with unmatched expertise. Our commitment to rigorous clinical operations ensures that every clinical performance study meets all regulatory standards, incorporating strategic risk management and adaptability for maximum compliance and integrity.
Partnering with us offers manufacturers a significant advantage, rigorously evaluating IVDs to ensure adequate performance and safety, a critical component of regulatory approvals.
Choose MDx CRO for excellence in IVD clinical performance studies, driving success and enhancing patient outcomes. Contact us for a discussion today!
Frequently Asked Questions about ISO 20916:2024 in 2026
Does ISO 20916:2024 apply to all IVD performance studies under IVDR?
ISO 20916:2024 provides a structured framework for the design, conduct, recording, and reporting of clinical performance studies involving IVD medical devices. While IVDR sets the legal requirements, ISO 20916 supports sponsors in demonstrating compliance through a harmonized and internationally recognized standard. Not all studies are identical in scope or risk level, but for interventional and other performance studies involving risk to subjects, alignment with ISO 20916 is strongly recommended to ensure methodological and documentation consistency.
What is the practical relevance of Annex ZA in ISO 20916:2024?
Annex ZA explains the relationship between ISO 20916:2024 and the requirements of IVDR. It maps the clauses of the standard to the corresponding IVDR provisions, helping sponsors understand how applying the standard supports regulatory compliance. In practice, Annex ZA serves as a bridge between operational study conduct and regulatory expectations under IVDR.
Why is ISO 20916:2024 strategically important for sponsors in 2026?
In 2026, regulatory scrutiny around clinical evidence and performance data continues to increase. ISO 20916:2024 offers a structured and prescriptive framework that reduces ambiguity in study design, monitoring, and documentation. For sponsors, early alignment with ISO 20916 can help minimize deficiencies during review, improve study robustness, and support smoother interactions with Notified Bodies.
Is ISO 20916:2024 mandatory under IVDR?
ISO 20916:2024 is not a regulation. IVDR is legally binding, whereas ISO 20916 is a standard. However, when recognized as harmonized, applying the standard provides a presumption of conformity with relevant IVDR requirements. Even where not mandatory, it is widely considered best practice for structuring clinical performance studies.
Written by:
Carlos Galamba
CEO
Senior regulatory leader and former BSI IVDR reviewer with deep experience in CE marking high-risk IVDs, companion diagnostics, and IVDR implementation.
The IVDR Shift and What It Means for Clinical Laboratories
The in Vitro Diagnostic Regulation (IVDR) (EU) 2017/746 came into force on 26 May 2022, representing a paradigm shift for diagnostic testing in Europe. Its purpose is clear: ensure safety, traceability, and performance of all in vitro diagnostic devices (IVDs). Unlike its predecessor, the IVDD (98/79/EC), the IVDR applies far-reaching obligations not only to manufacturers but also to clinical laboratories that develop and use their own in-house IVDs (IH-IVDs).
A cornerstone of this new landscape is Article 5(5), which sets conditions under which health institutions may continue manufacturing and using in-house devices without CE marking. While this exemption acknowledges the clinical need for tailored diagnostics, it also imposes new responsibilities.
This blog provides a step-by-step readiness checklist for laboratories to guide you through the transition.
Step 1: Do You Know What Counts as an In-House IVD?
What exactly is an in-house IVD under the IVDR?
An in-house IVD (sometimes called a laboratory-developed test or LDT) is any in vitro diagnostic device manufactured and used only within a health institution, not supplied to another legal entity, and not manufactured on an industrial scale
Examples include:
PCR assays where the lab develops its own probes.
Custom-developed software tools for diagnostic interpretation.
Excluded are:
General laboratory supplies.
RUO (research use only) products – unless repurposed for diagnostic use. If an RUO product is used for diagnostic purposes (i.e., results are communicated to the patient for medical decision-making), it ceases to be RUO and must comply with IVDR Article 5(5), thereby becoming subject to the same obligations as an in-house IVD/LDT.
Commercially available CE-marked IVDs (which must be purchased and used as intended) – unless it is modified, combined or used outside it’s intended purpose.
Takeaway:
You must determine whether you are using an in-house IVD. If you are modifying, combining, or using CE-marked diagnostic tests outside their intended purpose, or if you are repurposing RUO products for diagnostic use, you must ensure compliance with Article 5(5).
Step 2: Is Your Laboratory Recognized as a Health Institution?
Who is entitled to the Article 5(5) exemption?
Only health institutions may use in-house IVDs. According to the IVDR, a health institution is an organization whose primary purpose is patient care or public health. This includes:
Hospitals
Clinical laboratories
Public health institutes
Importantly, the recognition of health institutions may depend on national legislation. For instance, some countries require formal registration or accreditation to benefit from Article 5(5).
Takeaway:
Always check your national laws to confirm whether your laboratory qualifies as a “health institution” and whether additional national restrictions or obligations apply.
Step 3: CE-Marked or In-House? Make a Strategic Choice
Should your lab buy CE-marked tests or continue with in-house ones?
Under IVDR, labs face a strategic decision:
Purchase CE-marked IVDs: These carry regulatory assurance but may not always exist for niche diagnostic needs, and market withdrawals could limit supply.
Develop and use in-house IVDs: Allowed under Article 5(5) if your lab demonstrates compliance with conditions (e.g., GSPR, QMS, technical documentation).
From 31 December 2030, labs must justify why an equivalent CE-marked device is not suitable if they want to continue using their in-house test (article 5(5)(g))
Takeaway:
Begin analyzing your portfolio now. Which tests could be replaced by CE-IVDs, and which must remain in-house due to clinical need?
Step 4: Do You Comply with General Safety and Performance Requirements (GSPR)?
What technical documentation requirements already apply?
Since 26 May 2022, all in-house devices must comply with Annex I of the IVDR (GSPR). This includes:
Risk management system covering patient, user, and use error risks.
Performance evaluation based on scientific validity, analytical performance, and clinical performance.
Traceability and identification (lot numbers, production dates).
Appropriate instructions for use and safety information
Takeaway:
Treat your in-house tests with the same rigor as CE-marked devices. Maintain documentation to always prove compliance with the GSPRs.
Step 5: Have You Implemented an Appropriate QMS?
What does IVDR require for quality management when operating under article 5.5?
Since 26 May 2024, labs must manufacture and use in-house devices under an appropriate Quality Management System (QMS). For in-house IVDs, this generally means compliance with EN ISO 15189 or equivalent national provisions
However, note:
ISO 15189 covers quality in medical laboratories but not necessarily manufacturing processes.
Therefore, supplement with elements of ISO 13485 for design and production control.
In addition, laboratories must address the QMS requirements described in Article 10(8) IVDR, which outline the minimal aspects of a system covering risk management, manufacturing documentation, monitoring, corrective actions, and communication with authorities.
Takeaway:
Expand your QMS to cover risk management, manufacturing documentation, monitoring, and corrective actions, and the additional QMS obligations set out in Article 10 IVDR. Note that ISO 15189 alone is not sufficient; relevant elements of design and manufacturing from ISO 13485 must also be considered, as the IVDR introduces further QMS requirements that must be fulfilled.
Step 6: Can You Provide a Public Declaration?
Do labs need to publish information about their in-house devices?
Article 5(5)(f) IVDR requires health institutions to draw up and make publicly available a declaration for each in-house device. This obligation has applied since 26 May 2024, following the end of the initial transition period.
What must the declaration contain? At minimum:
Name and address of the health institution manufacturing the device.
Details necessary to identify the device (e.g., designation, type, internal code).
A declaration of compliance with Annex I (GSPR), or where full compliance is not possible, a reasoned justification explaining the deviations.
Confirmation that the device is manufactured under an appropriate QMS.
This declaration must be kept up to date and made easily accessible, typically via the laboratory or hospital’s website This transparency ensures accountability and facilitates oversight.
Takeaway:
Prepare standardized declarations for each in-house device. A practical tool exists: the IVDR Taskforce Guidance on LDTs (2020) provides a template (Appendix B) for the declaration that can be directly adapted by laboratories.
Step 7: Are You Ready for Competent Authority Oversight?
What role do regulators play?
Competent authorities may request documentation or even audit your lab to verify compliance. Labs must be prepared to show:
Design, manufacturing, and performance documentation of their in-house devices.
Clinical justification for developing or using the test instead of a CE-marked alternative.
Ongoing performance review and vigilance records, including corrective actions and monitoring of clinical use.
Evidence of an appropriate Quality Management System (QMS), as required since 26 May 2024.
The degree of oversight varies across Member States. For example, Belgium and Ireland already operate registration portals where laboratories must register their in-house tests. In other countries, legislation is still under development (Spain) or practices remain vague.
Takeaway:
Anticipate audits. Keep a compliance file for each in-house IVD.
Step 8: How Will You Justify Non-Equivalence? (2030 Requirement)
What happens in 2030?
From 31 December 2030, labs must justify why the specific needs of their target patient group cannot be met by a CE-marked device – Article 5(5)(g).
This justification may be based on:
Technical aspects (e.g., higher sensitivity).
Biological aspects (e.g., pediatric vs adult reference ranges).
Clinical needs (e.g., unmet diagnostic gaps).
Takeaway:
Start now by mapping your portfolio and identifying tests likely to face challenges in proving non-equivalence.
Step 9: Do You Have the Resources?
Why are many labs struggling?
Challenges highlighted in recent analyses include:
Lack of dedicated regulatory staff.
Limited time and budget for documentation.
Unfamiliarity with regulatory terminology.
Takeaway:
Seek structured support, whether through consultants, digital tools, or peer networks, to avoid non-compliance.
Step 10: Checklist. Is Your Lab Ready?
Step 1: Perform a GAP Assessment
Map your current situation: List all in-house IVDs and how they are used in your lab.
Check national status: Verify if your institution qualifies as a “health institution” under national law, and review whether national legislation imposes additional obligations such as mandatory QMS accreditation (e.g., ISO 15189), registration of in-house IVDs with competent authorities, or other reporting requirements that go beyond the IVDR.
Compare requirements vs. practice: Review the IVDR Article 5(5) obligations and identify where your lab already complies (e.g., risk management, traceability) and where gaps exist (e.g., QMS documentation, technical documentation).
Prioritize risks: Highlight critical areas (such as missing QMS procedures or incomplete Annex I documentation) that could block compliance in an inspection.
Step 2 – Take Action to Close the Gaps
Strategic choice: Decide whether to replace tests with CE-IVDs or maintain in-house versions. Document the rationale.
Annex I (GSPR): Ensure all in-house IVDs comply with General Safety and Performance Requirements (effective since 26 May 2022).
Quality Management System: Implement or update your QMS to align with ISO 15189, supplemented with elements from ISO 13485 and Article 10(8) IVDR.
Compliance documentation & oversight readiness: Compile and maintain a compliance file for each in-house IVD, including full technical documentation (design, manufacturing, risk management, and performance evaluation). Ensure these files are audit-read and can be provided upon request by competent authorities.
Vigilance & corrective actions: Set up procedures for monitoring performance, handling incidents, and implementing corrective/preventive measures.
Public declaration: Draft and publish a declaration for each in-house device (mandatory since 26 May 2024). Use available templates from guidance.
2030 justification: Start documenting why no equivalent CE-IVD meets the needs of your patient population to support continued in-house use after 31 December 2030.
Closing Thoughts
The IVDR sets high expectations for laboratory-developed in-house IVDs, transforming informal diagnostic practices into rigorously controlled processes. While compliance requires effort, resources, and cultural change, it also strengthens quality, safety, and patient trust. For laboratories, the transition is not optional, it is an opportunity to embed regulatory excellence into daily operations and secure the future of innovative diagnostics. Are you ready for the IVDR transition? Start today with a gap analysis, QMS reinforcement, and documentation plan. The earlier you act, the smoother your path to compliance will be.
At MDx CRO, we specialize in helping clinical laboratories navigate the IVDR, from gap assessments to QMS implementation and technical documentation. We support laboratories in demonstrating compliance with Article 5(5) for in-house IVDs by assisting with:
Gap assessments: Mapping all in-house IVDs, comparing current practice with IVDR Article 5(5) requirements, and identifying compliance gaps.
QMS alignment: Extending ISO 15189-based systems with manufacturing and design elements from ISO 13485, plus additional QMS obligations under IVDR.
Public declarations: Drafting and publishing Article 5(5)(f) declarations using recognized templates, ensuring accessibility and consistency.
Regulatory readiness: Preparing for competent authority oversight, including audits and requests for documentation.
Strategic portfolio decisions: Advising whether to replace tests with CE-IVDs or justify continued in-house use, including preparing 2030 equivalence justifications.
Vigilance systems: Setting up monitoring, incident reporting, and corrective/preventive actions in line with IVDR obligations.
Our team knows the pitfalls and the solutions. Let us support you in achieving full compliance. Contact us today to discuss how we can help.
Written by:
Hugo Leis, PhD
Training & Quality Manager
Quality & Training Manager and Senior IVDR consultant with expertise in CE marking, Clinical Laboratories, SaMD, Precision Medicine, Quality Assurance, and academic lecturing.
Impact of the Health Services Pack on IVD manufacturers, labs/health institutions and sponsors of combined studies
January 13, 2026
Carlos Galamba
In this article, we analyze the Health Services Pack IVDR impact and its role in shaping future health regulations.
On 16 December 2025, the European Commission published a proposalto amend the EU Medical Devices Regulation (MDR) and the In Vitro Diagnostic Regulation (IVDR) with targeted measures intended to reduce regulatory complexity, cost, and unpredictability while maintaining high safety standards.
This article focuses on IVDs under IVDR and the combined-study interface (drug–diagnostic and multi-legislation studies). It explains what the proposal says today, what it could change in practice, and how different stakeholders can prepare. It also recognizes that the text is still a proposal and may change during the ordinary legislative procedure in the European Parliament and Council.
Executive summary
What matters most for IVDR stakeholders.
If adopted largely as proposed, the package could materially affect how IVD stakeholders plan certification lifecycles, evidence generation, post-market obligations, in-house testing, and combined-study authorisations:
PRRC organisational burden would ease for SMEs relying on external PRRC support (availability requirement would soften; detailed qualification rules would be removed).
Certificate validity would shift from a fixed 5-year maximum to risk-based periodic reviews during the certificate lifecycle.
The proposal would support a broader evidence toolbox, including wider “clinical data” recognition and explicit promotion of New Approach Methodologies (including in silico testing).
Administrative burden would reduce via a narrower scope for summary documents and lower PSUR update frequency, plus longer timelines for certain vigilance reporting.
In-house testing under IVDR Article 5(5) would become more flexible, including (under IVDR) removing the “no equivalent device on the market” condition and explicitly bringing certain central laboratories supporting clinical trials into scope.
For combined studies, sponsors could submit a single application triggering a coordinated assessment aligned with the Clinical Trials Regulation framework.
Digitalisation would expand: digital EU Declarations of Conformity, more electronic submission, and electronic IFU for near-patient tests (among other measures).
The Commission frames these changes as a way to keep safety standards high while improving predictability, competitiveness, and innovation support; it cites €3–5 billion/year in cost savings at conservative estimates.
Key regulatory updates
PRRC flexibility for SMEs
Risk-based certificate validity
Broader clinical evidence
Reduced administrative burden
Flexible in-house testing (IVDR)
Streamlined combined studies
Expanded digitalisation
1) Context: why the Commission proposed a targeted IVDR/MDR revision
The Commission’s Q&A states that evaluation work identified shortcomings that negatively affect competitiveness, innovation, and patient care—pointing to inefficient coordination, divergent application of requirements, and procedures that are overly complex and costly.
The Commission describes the reform’s core objectives as:
Reduced administrative burden and stronger coordination
More proportionate requirements, especially for lower/medium risk devices and small patient populations
Support for innovation, including early expert advice and regulatory sandboxes
Greater predictability and cost-efficiency of certification, including enabling real-world evidence
Increased digitalisation across compliance tools and conformity assessment procedures
For IVDR stakeholders, the significance is not only the “what” but also the “how”: the proposal aims to make the system more predictable and less duplicative while leveraging EU-level expertise (including expert panels and EMA support).
2) Understanding Health Services Pack IVDR Impact: what it means for IVD manufacturers
2.1 PRRC: reduced organisational friction (especially for SMEs)
The proposal would:
Remove detailed qualification requirements for the Person Responsible for Regulatory Compliance (PRRC), and
Remove the requirement that SMEs using an external PRRC must have the PRRC “permanently and continuously” available; the PRRC would need to be available (without the “permanently and continuously” standard).
Why it matters: This could reduce structural overhead for smaller IVD manufacturers and for non-EU manufacturers using EU-based regulatory operating models. It may also reshape how manufacturers design PRRC coverage (internal vs external, shared services, outsourcing structures).
2.2 Certificates: from 5-year re-certification to risk-based periodic review
The proposal would remove the current maximum 5-year certificate validity and replace it with periodic reviews proportionate to device risk during the certificate’s validity period.
In addition, the proposal’s summary of certification changes includes:
Reduced systematic technical documentation assessment during surveillance activities (as summarised for certain IVD classes),
The ability for notified bodies to replace on-site audits with remote audits, and
Surveillance audits “only every two years” where justified by the absence of safety issues, plus unannounced audits “for-cause”.
Why it matters: This is a structural shift in compliance planning—from a calendar-driven re-certification event to an ongoing lifecycle model that could be more data-driven. IVD manufacturers will likely need stronger “always audit-ready” systems and clearer change-control strategies.
2.3 Evidence toolbox: broader clinical data concepts and explicit support for in silico approaches
The proposal summarises multiple evidence-related changes, including:
A wider range of data qualifying as clinical data,
More flexible conditions for relying on clinical data from an equivalent device, and
Promotion of New Approach Methodologies such as in silico testing.
Why it matters for IVDs: IVDR evidence expectations are often the pacing item for certification and market access—particularly for novel biomarkers, decentralised testing, and CDx. A broader toolbox could let manufacturers structure performance evaluation more efficiently, but it also puts more emphasis on robust justification: the proposal supports flexibility, not a “free pass.”
2.4 Summary documents and PSUR: targeted burden reduction
The proposal would:
Reduce the scope of devices that must have a summary of safety and (clinical) performance (SS(C)P) to those where the notified body must conduct technical documentation assessment—and remove the need for separate notified body validation of the draft summary.
Reduce the required PSUR update frequency, with notified body PSUR review integrated into surveillance.
For IVDR specifically, the proposal text also states that:
Manufacturers of class C and D devices would update the PSUR in the first year after the certificate is issued and every two years thereafter (or earlier in defined change situations).
Why it matters: This change could reduce recurring workload—yet it will likely increase expectations that PSUR content is meaningful, well-argued, and operationally integrated into surveillance interactions.
2.5 Vigilance and cybersecurity: longer timelines for certain incidents, plus cyber reporting alignment
The proposal would extend the reporting timeline for certain serious incidents (those not linked to public health threats, death, or serious deterioration) to 30 days instead of 15.
It would also introduce a cybersecurity linkage:
Certain MDR/IVDR vigilance reports that also qualify as actively exploited vulnerabilities or severe incidents under the cyberresilience framework would be made available to national CSIRTs and ENISA; and
Manufacturers would have to report actively exploited vulnerabilities and severe incidents that do not qualify as “serious incidents” under MDR/IVDR to CSIRTs and ENISA through Eudamed;
Cybersecurity would be explicitly mentioned in Annex I general safety and performance requirements.
Why it matters for IVD manufacturers: Cybersecurity is not only an “IT topic.” It increasingly affects performance, safety, vigilance, and field actions—especially for connected IVD instruments, software-driven diagnostics, and laboratory information system integration.
2.6 Digitalisation: eDoC, electronic submissions, eIFU for near-patient tests, and online sales information
The proposal includes:
Digital EU Declarations of Conformity,
More electronic submission of MDR/IVDR information,
Economic operators providing digital contacts in Eudamed,
Digital technical documentation and conformity assessment documentation, and
For IVDs, the ability for manufacturers of near-patient tests to provide electronic instructions for use.
It also introduces online-sales transparency requirements: essential device identification information and IFU information must be provided for online sales.
Why it matters: This points toward a compliance ecosystem where document control, traceability, and market surveillance become more data-centric. Manufacturers will need disciplined digital governance to prevent inconsistency across channels.
The proposal would make in-house conditions more flexible, including:
Allowing the transfer of in-house devices when justified by patient safety or public health interests, and
Under IVDR, removing the condition that no equivalent device exists on the market.
It also explicitly adds central laboratories manufacturing and using tests exclusively for clinical trials into the scope of the in-house device exemption.
Why Health Services Pack IVDR matters If adopted, this could significantly affect:
The role of hospital laboratories in innovation and continuity-of-care testing,
How health systems respond to unmet needs, niche populations, and rapidly evolving clinical practice, and
The operational models used to support clinical trials (including biomarker-driven trials and decentralised sample workflows).
What labs should plan Recognising the proposal may change
Governance and documentation systems that can withstand scrutiny as “in-house” use expands in scope and visibility.
Contracting and quality interfaces between health institutions, trial sponsors, and central labs—especially where a lab’s “in-house” position interacts with trial requirements and sponsor expectations.
4) What the proposal could change for sponsors of combined studies (drug–diagnostic interface)
4.1 A single application with coordinated assessment (CTR-aligned pathway)
For combined studies involving medicinal products, medical devices, and/or IVDs, the proposal states that a sponsor may submit a single application triggering a coordinated assessment in accordance with the Clinical Trials Regulation (CTR), noting alignment with amendments anticipated via the Biotech Act.
Why it matters: Sponsors running biomarker-driven programmes often experience friction at the interface between medicinal product trial authorisation processes and IVDR performance study requirements. A coordinated model—if implemented in a practical, predictable way—could materially improve planning across Member States.
4.2 Performance study burden reduction in defined scenarios
The proposal also states that:
Performance studies involving only routine blood draws would not require prior authorisation; and
Notification of performance studies on companion diagnostics using left-over specimens would be removed.
Why it matters: This could affect study-start timelines, especially in multi-country settings where administrative sequencing often drives critical path. For sponsors, it may also change how they design sample strategies, feasibility, and site activation planning.
5) Practical implications of Health Services Pack IVDR Impact: how stakeholders can prepare while the text remains a proposal
Because the proposal may change during negotiations, stakeholders should avoid “over-implementing” assumptions. At the same time, most organisations can act now in ways that remain valuable under multiple legislative outcomes.
5.1 For IVD manufacturers (RA/QA and clinical/performance teams)
Focus now on “no-regret” preparedness:
Map your portfolio to where the proposal signals the biggest change: certificate lifecycle management, audit model (remote/on-site), and surveillance cadence.
Re-evaluate your evidence strategy so it can flex across clinical studies, literature, equivalence, and (where applicable) in silico methodologies—while keeping scientific validity and traceability strong.
Strengthen change control to align with the proposal’s intent to distinguish changes that require different levels of notified body interaction (including predetermined change control planning).
Upgrade vigilance and cybersecurity workflows so reporting pathways align with both vigilance obligations and the proposed cyber reporting linkages (CSIRTs/ENISA/Eudamed).
Digitise with discipline: ensure eDoC, digital IFU strategies, and online-sales content controls remain consistent and auditable.
5.2 For labs and health institutions
Review how in-house governance could evolve if the “no equivalent device” condition disappears and trial-supporting central labs fall clearly within scope.
Align in-house test lifecycle controls with quality expectations likely to increase as in-house scope expands in visibility and operational relevance.
5.3 For sponsors of combined studies
Build study-start strategies around the proposal’s direction of travel: a coordinated route for combined studies and reduced administrative hurdles for defined performance study scenarios.
Stress-test protocols for evidence coherence: regulators will still expect sponsor claims and IVD performance claims to align, even if administrative routes simplify.
6) Health Services Pack IVDR Impact and its potential global reach: what this could mean outside Europe
The Commission positions the EU as a global leader in medical device regulation and indicates the reform aims to make the sector more competitive globally. It also explicitly links the proposal to reinforcing international cooperation, including participation in high-standard international cooperation and information-sharing mechanisms with reliable partners and strengthened uptake of international guidance.
For global manufacturers, that matters because EU compliance strategies often influence:
Global clinical evidence planning and dossier structuring, and
How manufacturers operationalise post-market surveillance and cybersecurity controls across regions.
(How much convergence happens in practice will depend on implementation and on how reliance mechanisms are used over time.)
FAQs
Is the Health Services Pack already law?
No. The Commission published a proposal on 16 December 2025. The text must go through the ordinary legislative procedure in the European Parliament and Council before any final legal changes take effect.
Will the proposal change IVDR certificate validity?
The proposal would remove the maximum 5-year certificate validity and replace it with risk-based periodic reviews while the certificate remains valid.
Does the proposal reduce PSUR update frequency under IVDR?
Yes. The proposal would reduce PSUR update frequency and integrate notified body PSUR review into surveillance. It also states class C and D PSUR updates would occur in the first year after certification and every two years thereafter (or earlier in defined cases).
Does the proposal change serious incident reporting timelines under IVDR?
Yes. For serious incidents not related to public health threats, death, or serious deterioration, the proposal would extend reporting timelines to 30 days instead of 15.
Does the proposal change IVDR in-house testing rules?
Yes. The proposal would make in-house conditions more flexible and, under IVDR, would remove the condition requiring “no equivalent device on the market.” It would also add certain central laboratories supporting clinical trials into scope.
How would the proposal affect combined studies?
The proposal states that sponsors could submit a single application for combined studies, triggering a coordinated assessment aligned with the Clinical Trials Regulation framework.
How can MDx CRO help you navigate Health Services Pack IVDR Impact effectively?
If you manufacture IVDs, run laboratory services, or sponsor combined studies, you will likely need to translate the proposal into:
A portfolio-level impact assessment (technical documentation, evidence strategy, and certification lifecycle planning), and
An operational plan for performance studies and combined-study submissions that remains robust even if the final text changes.
MDx CRO supports IVD manufacturers and sponsors across IVDR technical documentation, performance evaluation strategy, and combined study operational delivery (clinical operations + RA/QA alignment). The most effective next step is usually a short, structured gap-and-opportunity review tied to your portfolio and pipeline.
Need support?
We can assist you translating the Health Services Pack proposal into practical IVDR actions for your portfolio, studies, or lab activities.
Senior regulatory leader and former BSI IVDR reviewer with deep experience in CE marking high-risk IVDs, companion diagnostics, and IVDR implementation.
Gain access to MDx’s Precision Medicine Services Pack—detailing our capabilities in clinical research, regulatory strategy, and companion diagnostics. Learn how we support pharma, CDx, and advanced therapy developers across the clinical and regulatory lifecycle.