In an era where digital health, telemedicine, and AI-driven diagnostics are becoming mainstream, Software as a Medical Device (SaMD) is no longer a niche concept; it stands as a core pillar of modern healthcare innovation. Yet, delivering a safe, effective, and compliant SaMD product in Europe requires careful navigation of complex regulatory regimes.
For companies and regulatory affairs teams, successful market access in the European Union means meeting the demands of the EU Medical Device Regulation (MDR, Regulation (EU) 2017/745) and, increasingly, the EU Artificial Intelligence Act (AI Act, Regulation (EU) 2024/1689). Together, this combined regime shapes how developers design, validate, monitor, and maintain software with medical functionality.
This SaMD Compliance Guide presents a concise, European-focused overview. You’ll find:
- How to determine if your software qualifies as SaMD
- Key requirements under the MDR (classification, conformity, clinical evaluation, post-market)
- Best practices, pitfalls, and strategic recommendations
1. Defining SaMD: What Qualifies?
What is SaMD?
The International Medical Device Regulators Forum (IMDRF) defines SaMD as:
“Software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.”
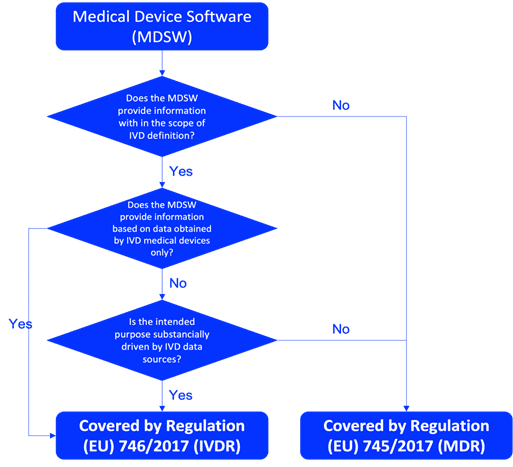
In the EU context, and based on MDCG 2019-11, software qualifies as a medical device when the manufacturer’s intended purpose includes diagnosis, prevention, monitoring, prediction, prognosis, treatment, or alleviation of disease.
Key determinants
- Intended medical function (not administrative, not purely wellness)
- Standalone operation; the software does not need to embed in medical hardware
- Potentially autonomous action (e.g., cloud-based analysis)
Examples (and non-examples)
Typical SaMD examples
- An AI-based image analysis tool that assists radiologists in detecting tumors
- A mobile app that predicts hypoglycemic events for diabetic patients
- A cloud algorithm that classifies ECG signals to detect arrhythmias
Non-SaMD (or out-of-scope) software
- A healthcare facility’s scheduling or billing software
- A fitness tracker app for general wellness (unless marketed for disease diagnosis)
- A general-purpose image viewer used in the clinic but not intended for diagnosis
Because the line can be subtle, regulatory teams should document a short justification for whether software is—or is not—a medical device, supported by functional claims, labeling, and architecture.

2. The EU MDR Framework for SaMD
Classification: Rule 11 for Software
Annex VIII of MDR includes Rule 11, which addresses software risk classification. Under Rule 11:
- If the software informs decisions for diagnostic or therapeutic purposes, it often lands in Class IIa, IIb, or even Class III, depending on risk and the consequences of error.
- If the software monitors physiological processes, it may fall in Class IIa or IIb.
- Software intended for administrative or non-medical functions typically falls in Class I.
Because many advanced SaMD tools now trigger Notified Body oversight, developers should plan conformity assessments, clinical evaluation, and documentation accordingly.
The MDR Compliance Roadmap
To achieve CE marking under MDR, follow these essential steps:
- Intended Purpose & Use Context – Define the intended medical purpose, user groups, environment, contraindications, and usage scenarios with precision.
- Risk Management (ISO 14971) – Identify hazards and mitigate risks, including software bugs, algorithm drift, cybersecurity intrusion, and data errors. Manage risk across the full lifecycle (design, validation, deployment, maintenance).
- Quality Management (ISO 13485) – Operate under a QMS that addresses design control, configuration management, change control, CAPA, and supplier management.
- Software Lifecycle (IEC 62304 / 82304-1) – Use recognized lifecycle standards to structure architecture, module-level design, verification and validation, maintenance, and configuration.
- Clinical Evaluation (MDCG 2020-1) – Demonstrate clinical benefit and performance with fit-for-purpose evidence.
- Technical Documentation (Annex II/III) – Include architecture, risk analysis, verification, usability, labeling, and performance claims.
- Conformity Assessment – For Class I(s/m/r), IIa and above, a Notified Body reviews your QMS and technical documentation and performs audits.
- CE Marking & Declaration of Conformity – Once you demonstrate conformity, apply the CE mark and sign the DoC to enter the EU market.
- Post-Market Surveillance – Maintain PMS and PSUR, and integrate performance data and AI monitoring logs.
- Software Updates and Change Control – Analyze each change—functional, algorithmic, or data-driven—to decide whether it represents a significant change that requires re-assessment.
3. Cybersecurity and Lifecycle Protection
Cybersecurity should start at design and continue through maintenance. The main requirements include:
- Ensure confidentiality, integrity, and availability (CIA) throughout the lifecycle
- Define minimum IT requirements and secure configurations
- Implement verification and validation of security controls
- Provide clear IFU instructions on data protection, updates, and secure disposal (GSPR 13.6)
- Maintain a post-market security plan to track vulnerabilities and manage patches
4. Challenges, Risks & Strategic Recommendations
| Challenge | Mitigation / Best Practice |
|---|---|
| Unclear intended purpose or software classification | Define the medical purpose at project initiation. Align IFU, labeling, marketing, and technical files with intended use and Rule 11 logic. |
| Insufficient clinical/performance evidence | Use prospective studies or robust real-world performance evaluations aligned with MDR Annex XIV and, where applicable, AI Act testing provisions. |
| Data quality and representativeness | Implement data governance for acquisition, preprocessing, and validation. Ensure datasets represent the intended patient population and use context. |
| Transparency and user comprehension | Provide clinically interpretable outputs. Explain functionality, limitations, and user responsibilities in the IFU and training materials. |
| Traceability gaps between requirements, risks, and tests | Maintain a requirements-to-verification traceability matrix that links requirements, risk controls, verification results, and clinical claims. |
| Software updates and regulatory impact | Establish change management to evaluate whether updates are significant and require re-assessment. Integrate these controls into the QMS. |
| Regulatory and Notified Body capacity constraints | Engage early with a qualified Notified Body. Provide clear, harmonized documentation to streamline assessments. |
| Evolving standards and regulatory guidance | Monitor new EU and MDCG guidance and standards (ISO 14971, ISO 13485, IEC 62304, IEC 81001-5-1) and the EU AI Act. Review QMS procedures periodically to stay aligned. |
5. Conclusion
Delivering safe and compliant Software as a Medical Device (SaMD) requires a structured approach that integrates regulatory, technical, and quality considerations across the lifecycle. Compliance with the EU MDR ensures that safety, performance, and clinical benefit remain clear and consistently supported.
Advanced technologies, including AI, can enhance SaMD functionality; however, they should not overshadow the core principles of safety, effectiveness, and human oversight. The same regulatory rigor and lifecycle management practices apply to all SaMD, regardless of the underlying technology.
Manufacturers should:
- Define a clear intended purpose aligned with clinical benefit
- Maintain a QMS that addresses MDR and, where relevant, AI Act obligations
- Engage early with Notified Bodies and keep documentation, risk, and cybersecurity controls consistent
- Treat post-market surveillance and maintenance as continuous improvement
By embedding these principles, manufacturers can reach compliance efficiently and deliver trustworthy, clinically valuable SaMD solutions.