Case Study: How MDx Helped Fulgent Genetics Achieve CE Marking for One of the World’s Largest Germline NGS Panels under IVDR
Outcome: CE mark granted by TÜV SÜD for an end-to-end Class C germline NGS solution (wet lab + bioinformatics).
Scope: Furthermore, panel covering 4,600+ clinically relevant genes with a validated PLM (Pipeline Manager) software component; later expanded to >7,000 genes using a new probe set.
What we did: Specifically, we built an ISO 13485 QMS from the ground up, prepared full Annex II + III technical documentation, validated bioinformatics under IEC 62304/82304, split documentation into two Basic UDI-DIs (wet lab vs. software), and guided Stage I/II audits.
Why it matters: Ultimately, this demonstrates a repeatable pathway to IVDR certification for large NGS services and software—something that hadno clear precedent.
The challenge: certifying a service-based, large-scale NGS system under IVDR
To begin with, FulgentExome is a service-based NGS solution that integrates wet-lab workflows with the Fulgent PLM bioinformatics pipeline. As a result, pursuing IVDR certification meant converting a mature CLIA/CAP testing service into a CE-marked IVD system with robust evidence across scientific validity, analytical performance, and clinical performance—for thousands of genes. In particular, key hurdles included: defining intended use at scale; validating third-party components; proving scientific validity across 4,600+ genes; above all fully validating the bioinformatics pipeline under medical device software standards.
MDx approach: a playbook for complex NGS + software
1) Build the right QMS, fast
First, we performed an IVDR GAP assessment. Next, we designed and implemented an ISO 13485-compliant QMS with risk management, supplier control, document control, internal audits, and management review—migrating from a CLIA/CAP model to IVDR-ready operations.
2) Engineer a defensible intended use
Meanwhile, the intended-use statement evolved iteratively—from an initial ~300-gene scope to whole-exome, finally landing on 4,600+ genes aligned to available clinical and analytical evidence. In the end, the final language was future-proofed to support rapid updates as evidence expands.
3) Split wet lab and software into two regulated products
Afterward, following round 1 review feedback, we separated the documentation into two Basic UDI-DIs—FulgentExome (wet lab) and Fulgent PLM (software)—to align with IVDR expectations for traceability and lifecycle control. Consequently, this restructuring sharpened conformity assessment and accelerated subsequent approvals.
4) Validate the informatics stack like a medical device
In parallel, we validated PLM under IEC 62304/82304, including architecture, version control, cybersecurity, verification/validation, and integration with external databases. Therefore, the result was a fully evidence-backed bioinformatics pipeline capable of reproducible, high-confidence variant calling and reporting.
5) Make “evidence at scale” practical
First,Scientific validity: Three-tier strategy combining validation of exome sequencing as an approach, reliance on curated public databases, and deep exemplars for a large subset of genes.
Second,Clinical performance: Leveraged routine testing experience (thousands of positives) to focus clinical evidence on high-prevalence genes, and instituted a robust PMPF strategy to continuously strengthen low-prevalence areas.
6) Orchestrate TÜV SÜD audits to success
Initially, Stage I confirmed documentation readiness, scope, Basic UDI-DIs and integration of IVDR processes into daily practice.
Subsequently, Stage II verified on-the-floor implementation of SOPs, training, competence, CAPA and change control—closing findings on short cycles to hit NB timelines.
Results that move the market
CE mark granted for FulgentExome & Fulgent PLM—among the first end-to-end Class C germline NGS solutions under IVDR.
Certified scope covers 4,600+ genes, positioning Fulgent as a reference lab for comprehensive hereditary disease testing serving European patients.
Post-certification, the platform scaled to >7,000 genes using a new probe set—demonstrating the inherent scalability built into the certified system (process, documentation, and change control).
Strengthened competitive standing in the EU diagnostics market; public communications highlight the magnitude of this achievement for large NGS panels.
What this means for labs and IVD developers planning large NGS submissions
If you operate an LDT today: you’ll need to translate CLIA/15189 practices into an ISO 13485 framework, document design controls, and produce a full PER (PEP/PER), APR, SVR, PMS/PMPF, SSP, and labeling/IFU aligned to GSPR. Expect to validate any bioinformatics pipeline as SaMD with IEC 62304/82304 and cybersecurity controls.
If your panel is “large”: you likely won’t have uniform clinical data across every gene. A structured tiered evidence model + PMPF can satisfy Notified Bodies while keeping your roadmap scalable.
If you combine wet lab + software: plan for separate Basic UDI-DIs and documentation sets. Treat the pipeline as a product with its own requirements, verification, and risk controls.
Why MDx
End-to-end IVDR expertise: From regulatory strategy & classification to Annex II/III technical files, PER/SVR/APR, training, and mock NB reviews.
Clinical performance studies: We design, run, and report ISO 20916 studies (protocols, eTMF, monitoring, biostats, PER alignment), and we can act as delegated sponsor for multi-country submissions—100% submission success rate to date.
Operational scale: ISO 9001 clinical QMS, EU/US partner network, multilingual CRAs, and a repeatable process honed on 60+ performance study submissions for top IVD and pharma clients.
Project timeline
Our joint project with Fulgent spanned July 2023–July 2025, with overlapping tracks for QMS creation, technical documentation, NB review, and Stage I/II audits—a coordinated plan that allowed rapid closure of findings and post-certification scaling.
Client perspective
The program demanded evening/weekend execution across regulatory, documentation, and project management to meet Notified Body timelines—effort that, in the client’s words, “made all the difference” in achieving what initially “seemed almost impossible.”
Planning IVDR for your NGS panel? Here’s a quick readiness checklist
Intended use aligned to evidence (and future updates)
ISO 13485 QMS with software lifecycle integration
PER (PEP/PER), SVR, APR mapped to gene-level strategy
PLM/DR pipeline validated per IEC 62304/82304 (+cybersecurity)
Separate documentation/UDI for wet lab vs. software (if applicable)
PMS/PMPF plan to mature low-prevalence evidence post-market
Mock NB review + Stage I/II audit readiness
(Our team can lead or co-author each artifact above.)
Talk to us
Whether you’re certifying a focused oncology panel or pushing the limits with exome-scale content, MDx brings the cross-functional regulatory, clinical, quality, and software depth to make it possible—on a timeline that keeps your business competitive.
Senior regulatory leader and former BSI IVDR reviewer with deep experience in CE marking high-risk IVDs, companion diagnostics, and IVDR implementation.
IVD Software Development: How to Bring IVD Software to Market in 8 Steps
April 4, 2023
The healthcare industry is undergoing a rapid transformation spurred by the advent of advanced medical diagnostic technology. IVD software development is a critical component of this revolution as it allows for testing, analysis, reporting, and communication without needing a physical laboratory or a visit to a doctor’s office.
Bringing IVD software development to the market can benefit patients and healthcare providers who can deliver quality care faster with fewer resources.
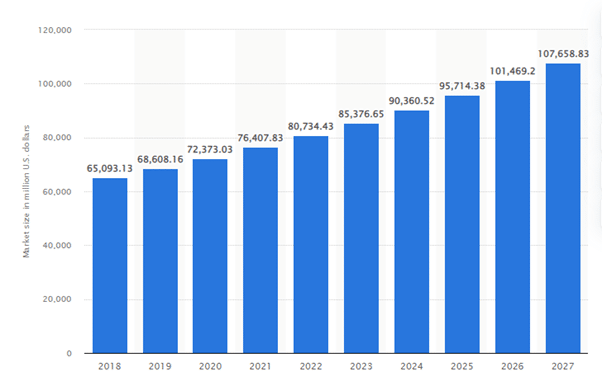
Projected size of the IVD market worldwide from 2018 to 2027 (in million U.S. dollars)
The above graph shows the In-Vitro Diagnostics (IVD) market globally was estimated at 72.4 billion U.S. dollars in 2020, with a projected growth of 108 billion U.S. Dollars by 2027, showing its increased relevance in the healthcare industry today.
1. Conduct Market Research
Before starting the development process, organizations must conduct market research and understand their target market, user needs, and potential competitors.
Market research should also help determine regulatory requirements that organizations must comply with and any current trends within the industry or technology space.
Below are some key points to consider when conducting market research for IVD software development:
Identify regulatory requirements: Identifying the regulatory requirements for your software is essential for bringing your product to market. They may vary depending on your target market, such as the FDA in the US or the European Union’s CE marking requirements (IVDR 746/2017).
Determine your target market: Identify the segments of the healthcare industry that your IVD software will serve. Consider factors such as geography, type of healthcare organization, and specialty areas.
Identify your competitors: Research the IVD software market to identify your competitors and their products. Analyze their strengths, weaknesses, pricing, and marketing strategies.
Understand your customers: Conduct surveys, interviews, and focus groups with healthcare professionals to understand their needs, preferences, and pain points. Use this information to tailor your IVD software to meet their specific needs.
Analyze market trends: Stay up-to-date on the latest trends and developments in the IVD software market. Monitor industry publications, attend conferences, and follow industry experts and thought leaders on social media.
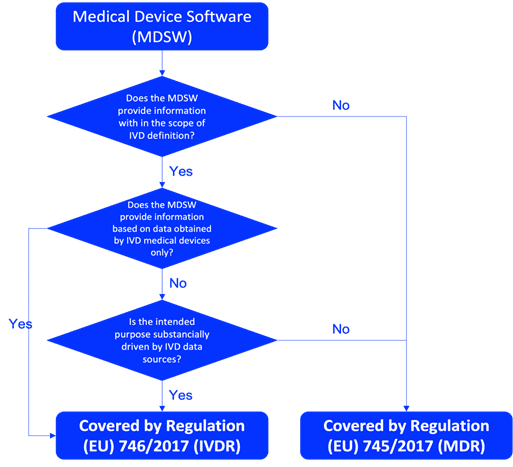
Understanding the classification of your IVD software is crucial before starting the planning and design process. Determine whether the IVD software is a standalone IVD medical device or a component of a larger system. To be qualified as an IVD medical device software in the EU, the product must fulfil the definition of IVD according to Article 2(2) of Regulation IVDR746/2017, as described in the Figure 1. The correct classification of IVD software should be done based on the rules described in Annex VIII of the IVDR. Relevant guidance documents, such as MDCG 2019-11 are also essentials for the qualification and classification of IVD software.
Fig. 1 – MDCG 2019-11 flowchart on qualification of Medical Device Software (MDSW)
Familiarize yourself with relevant regulatory frameworks, guidances and standards such as ISO 13485, IEC 62304, but also specific guidance documents published by regulators, which provide specifications and guidelines for developing, validating, and maintaining IVD software.
3. Plan and Design the Software
The next crucial step of a successful IVD software development is design and planning.
A well-documented and robust planning process can help provide a more detailed roadmap for development.
During this phase, design reviews, testing, and verification will ensure that the final version meets user requirements.
It is essential to incorporate user feedback at every stage of the design process to develop an intuitive interface that works effectively according to their needs.
Obtaining feedback from customers and stakeholders offers the development team opportunities to recognize potential concerns and areas for enhancement.
Developers can devise software solutions that fulfill customer needs and address their grievances by integrating feedback.
The importance of accurate documentation should not be underestimated as it helps trace back issues later on in the lifecycle of the software.
The development team must consider scalability and flexibility during the initial planning and design stages when creating the software.
4. Develop and Test the Software
Developing and testing the software is crucial in creating a working prototype.
The development phase is necessary to ensure the accuracy of the design, coding, and algorithms used in creating the software solutions.
Testing and quality assurance also play an essential role in ensuring the products meet all requirements before launch.
It is essential for companies to thoroughly assess each component of their software as part of this process. This includes ensuring they meet performance objectives concerning speed, responsiveness, scalability, security, reliability, and ease of use for their users.
Quality assurance checks help identify bugs or errors to release a defect-free product that meets all standards from regulatory bodies such as FDA or CE Marking.
When deploying IVD systems, manufacturers need to consider if their applications can be flexible enough to support new technological advances; future-proofing their products becomes increasingly necessary where customers demand longevity across upgrades or iterations over time.
5. Prepare a Regulatory Submission
Preparing a regulatory submission package is critical in bringing IVD software development to market. This step involves compiling documents to demonstrate that the software meets regulatory requirements and is safe and effective.
Here are some critical considerations for preparing a regulatory submission:
Gather relevant documents for the package
Understand regulations, standards, and risk classification of IVD software and the manufacturing role. Key documents to include are the device description, technical documentation, risk management file (ISO 14971), software lifecycle documentation (IEC 62304), and quality management system documentation (ISO 13485). Risk management must be applied and monitored during the IVD software development life cycle.
Prepare a performance evaluation report (PER)
This will require a comprehensive analysis of scientific evidence showing that the product meets user needs safely and effectively. IVD software performance evaluation should be prepared in accordance with relevant guidance documents, such as MDCG 2020-1 for the EU. Other guidance such as MedTech Europe Clinical Evidence Requirements for IVD can also be a good source of additional information.
Clinical performance studies are aimed at providing evidence of the safety and effectiveness of a product’s intended purpose to ensure that it’s able to diagnose, monitor and predict diseases and conditions accurately.
As described in MDCG 2020-1“Validation of the clinical performance should be considered at each change of the software to a new release. If no validation is performed, a justification should be stated in the technical documentation. With a validation of clinical performance, it is demonstrated that users can achieve clinically relevant outputs through predictable and reliable use of the MDSW”.
Adherence to relevant standards and guidelines, such as ISO 20916 (Clinical Performance Studies for In vitro diagnostics) and Good Clinical Practice (CGP), are crucial for the successful execution of clinical studies.
Ensure data accuracy
Ensure that that any data collected from testing is presented accurately to prove safety and efficacy before submitting your application. This includes validation and verification data, performance evaluations, and, if applicable, results from clinical studies. Carefully review all information for accuracy and completeness before submitting it.
6. Obtain Regulatory Approval
You need to obtain regulatory approval to bring IVD software development to market. Successful approval enables you to market and sell your software in compliance with local laws and regulations.
Familiarize yourself with the applicable regulatory frameworks and guidelines for your software. These may include ISO 13485 (quality management systems), IEC 62304 (medical device software lifecycle processes), ISO 14971 (risk management), and MDCG guidance documents such as MDCG 2019-11 (qualification and classification of software in the medical device regulations) and MDCG 2020-1 (guidance on performance evaluation for IVD software under the IVDR. You should also consider guidelines from other jurisdictions depending on your market strategy. The FDA for example has issued guidance for software as a medical device (SAMD).
Develop a comprehensive application package that should include all the necessary documents, data, and tests required for review. Ensure your package addresses regulatory requirements, such as conformity assessment procedures, clinical evidence, and post-market surveillance.
Submit the application package to the relevant regulatory authorities for review, such as the FDA in the US or notified bodies in the EU under the In Vitro Diagnostic Regulation (IVDR) 2017/746.
Be prepared to respond quickly and accurately to any feedback or additional information requests from regulatory agencies during the review process.
Make recommended changes swiftly as part of your submission to obtain approval from agencies successfully.
Once you receive approval from regulatory agencies, you can move forward with marketing and selling your software according to local laws & regulations, ensuring ongoing compliance with any post-market requirements.
7. Developing Marketing and Sales Strategies
Creating a successful marketing and sales strategy is essential for bringing IVD software to the market, it allows for faster positioning and gaining a competitive advantage. Make sure to develop a strong brand identity with messaging that resonates with your audience.
In addition, researching customer needs and understanding key industry trends can create a more targeted approach when it comes to the marketing of IVD software solutions, increasing your likelihood for success.
Make sure to use multiple channels such as paid advertising, email campaigns, social media and webinars to reach out to potential customers from diverse segments.
And last but not least, creating effective communication strategies to engage with customers throughout the sales cycle will also be key to promoting IVD products successfully.
8. Launch and Support the Software
Launching and supporting software is a crucial element to its success. The product can be improved over time by providing regular updates and customer service, and users can get the best experience.
Here are some points to consider when launching your IVD software development:
Create a comprehensive support plan that puts customer needs first. Ensure you have an efficient process for handling inquiries and technical issues as they arise.
Ensure that all necessary software updates are completed on schedule, so users don’t experience any delay in accessing the product’s full features or bug fixes.
Assess the regulatory impact of changes and bug fixes, changes to your IVD software may or may not be significant. Consider whether changes impact your regulatory approvals. MDCG 2022-6 provides additional guidance on changes to design and intended purpose in the context of the new transition timelines for IVDs in Europe.
Set up user feedback forms or surveys so customers can share their thoughts on the product’s performance and what improvements they want to see. This will help drive further development of the software over time.
Offer ongoing training opportunities for new features, so users feel confident using them once released. This will also ensure that customers know how to use their investment in your IVD software development solution fully.
Revolutionizing Healthcare With IVD Software Development
In vitro diagnostic (IVD) software development has transformed the healthcare industry by providing cost-effective testing, analysis, reporting, and communication solutions without physical laboratory equipment.
Studies indicate that healthcare providers highly prioritize in vitro diagnostic (IVD) procedures, and their optimization has the potential to enhance patient outcomes. Therefore, the development of IVD software is crucial in facilitating quicker and more accurate diagnostic results, ultimately leading to the optimization of healthcare practices.
If you need a partner in IVD software development for your business, MDx CRO is an IVD consultancy that provides end-to-end solutions to accompany you at each step of the process. Our team of highly experienced CRO strategists has extensive expertise in bringing innovative medical devices and IVD technologies to market. Request your expert consultation today.
FAQs
What are the key considerations when designing IVD software?
There are several key considerations that companies should keep in mind when designing IVD software: user requirements, regulatory requirements depending on the target geographic location, data accuracy and effective data management, the software’s ability to integrate with other systems, as well as performance and usability.
What are the regulatory requirements for IVD software development in Europe?
The regulatory requirements for IVD software development in Europe are determined by the In Vitro Diagnostic Regulation (IVDR), which became applicable on May 26, 2022. They include, but are not limited to design and development, risk management, validation and verification, as well as compliance with GDPR.
What are the most common challenges in IVD software development?
The most common challenges in IVD software development include regulatory compliance (which can be complex and challenging to navigate through), ensuring integration compatibility with other systems, effective data management, and great user experience, among others.
How do you ensure the quality and reliability of IVD software?
To ensure the quality and reliability of IVD software, it’s important that companies follow all regulatory guidelines applicable to their geographical location, and use a quality management system to ensure that the development process is well-documented. Conducting testing, validation and verification processes is another essential element of software development for in vitro diagnostics.
Gain access to MDx’s Precision Medicine Services Pack—detailing our capabilities in clinical research, regulatory strategy, and companion diagnostics. Learn how we support pharma, CDx, and advanced therapy developers across the clinical and regulatory lifecycle.