Combined studies, clinical trials that simultaneously investigate a medicinal product and an IVD under both the Clinical Trials Regulation (CTR) and the IVDR, are among the most complex regulatory challenges in precision medicine today. The lack of a coordinated EU assessment means sponsors face separate national submissions, divergent NCA interpretations, and timelines that can stretch 6-12 months beyond what was planned.
Last week, I presented at the 16th Clinical Biomarkers & Companion Diagnostics Summit Europe in London, sharing data and lessons from over 40 combined programs that MDx CRO has managed across 20+ EU countries. This article distils the key findings from that presentation: what triggers IVDR performance study requirements, why the same protocol can get three different answers from three NCAs, the top RFIs we see across programs, and how the regulatory landscape is evolving with MDCG 2025-5 and the December 2025 Health Services Pack.
Whether you are a pharmaceutical sponsor planning your first combined study, or a regulatory affairs professional navigating the dual-track CTR/IVDR submission process, this article gives you the operational intelligence that no guidance document provides.
Want the full CDx presentation slides?
Download the complete deck with case studies, the PSA approval timeline map, and the wave planning framework below.
Free download, presented at the 16th Clinical Biomarkers & CDx Summit Europe, London, March 2026
The Combined Studies Bottleneck: Why This Matters Now
The numbers tell the story. According to the EU COMBINE Analysis Report 2024, there were 402 combined study applications across the EU, of which 343 were CTR/IVDR combinations, and 86% of those were multinational. The mean approval time was 137 days, but the range was enormous: 45 to 267 days depending on the country, the submission pathway, and the quality of the initial dossier.
The root cause is structural. There is no coordinated IVDR assessment across Member States. Each country evaluates the performance study component independently, using national portals, national forms, and critically, national interpretations of the same regulation. For sponsors running multinational oncology trials with a companion diagnostic, this means multiplicative complexity: every additional country adds not just a submission, but a potentially different regulatory conclusion about whether a Performance Study Application (PSA) is even required.
And the volume is growing. Industry estimates project over 3,000 IVD submissions expected by 2029 as more CDx-driven oncology programs reach the EU. The system is already under strain.
This is the environment in which every combined study sponsor now operates. The question is not whether you will encounter divergent NCA opinions, it is how you prepare for them.
Same protocol, same device, three different answers.
The case presented at CDx Europe involved a pharmaceutical sponsor planning a multinational Phase 1b/2 oncology trial across France, Germany, and Spain. The study used a CE-marked companion diagnostic to run central testing retrospectively on leftover tumour samples, after enrolment was complete, purely as exploratory biomarker analysis. No additional invasive procedures. No impact on protocol-mandated treatment decisions. The sponsor sought pre-submission advice from three Member State competent authorities, presenting the same protocol and the same scientific rationale to each.
The three responses were incompatible.
NCA 1 concluded that no PSA was required, it accepted the sponsor’s argument that the activity was purely exploratory and non-interventional.
NCA 2 took the opposite position: because the assay was being used outside the scope of its CE-marked intended purpose, and because discrepant results would be communicated to investigators who could potentially act on them, it classified the study as interventional and required full PSA authorisation.
NCA 3 had not issued a definitive answer within the consultation period. This authority lacks a formal structured consultation procedure for performance studies and typically responds by email without fixed timelines.
When we presented this at the conference, there was an audible reaction in the room. Several attendees nodded immediately, this clearly resonated with their own experience. The most revealing question came from a regulatory affairs director at a mid-size pharmaceutical company who asked: “If the strictest authority changes its mind on appeal and agrees with the others, do we then need to explain to the permissive authority why we did something they told us we didn’t need to do?”
That question captures the operational absurdity of the current system perfectly. Sponsors are caught between harmonising upward, following the strictest requirement everywhere, which creates unnecessary burden in permissive jurisdictions, and harmonising downward, which creates compliance risk in the strictest one.
How Was It Resolved?
No harmonised consensus was reached between the three authorities. The sponsor appealed to the strictest authority in late 2025, providing additional justification and citing the fact that the most permissive authority had accepted the same protocol without any submission. By early 2026, the sponsor was cautiously optimistic that the appeal would succeed.
In parallel, they prepared contingency documentation for a dual-track approach: a full PSA in the country requiring it, a Performance Study Notification (PSN) in the country where the outcome remained uncertain, and no submission in the country that had cleared the protocol outright. Each country would effectively follow its own regulatory requirement.
Timeline impact: The divergent opinions introduced approximately 4–6 months of delay to the diagnostic component of the trial, encompassing multiple pre-submission consultation rounds, country-specific dossier preparation, appeal proceedings, and the need to compile and translate Annex XIV documentation for each jurisdiction independently.
There is currently no formal escalation or conflict-resolution pathway for performance study assessments. This is precisely the structural gap the COMBINE programme’s coordinated assessment pilot is intended to address.
What Triggers IVDR Performance Study Requirements?
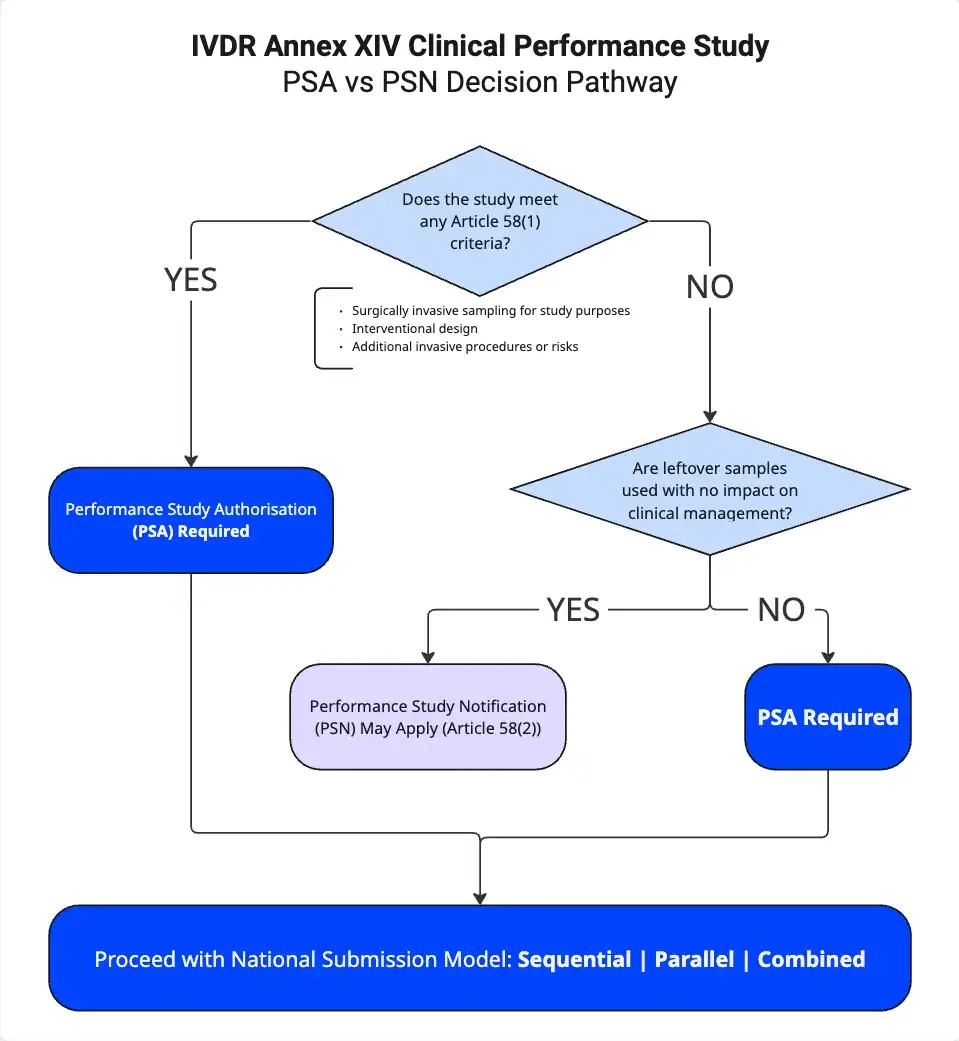
Understanding the regulatory pathways is critical for any combined study. The IVDR establishes three routes, and the distinction between them determines everything: your timeline, your documentation burden, and whether your study can start at all.
Article 58.1 Authorisation Required
This applies to non-CE-marked devices or devices used outside their intended purpose. The process involves a 25-day validation phase followed by a 45-day review. You need full PSA authorisation when the study involves surgically invasive sample-taking for study purposes only, when it is an interventional clinical performance study, or when there are additional invasive procedures or risks beyond standard care.
Article 58.2 Notification Pathway
This applies to companion diagnostics using leftover samples only, where results do not influence treatment during the trial. The pathway is immediate or NCA-dependent after validation, depending on the Member State.
Article 70 CE-Marked Devices
This is a 30-day notification route for CE-marked devices used within their intended purpose but involving additional invasive or burdensome procedures beyond normal use. A critical nuance: if a CE-marked IVD is studied outside its intended purpose, the Article 70 notification route does not apply, it must be assessed under Article 58 instead.
The Key Decision Factors
From our experience across 40+ combined programs, the questions that determine which pathway applies and where NCAs diverge, are consistent:
- CE mark status: for the specific trial use (intended purpose) not just whether the device has a CE mark, but whether the trial use falls within the marked scope
- Whether results impact medical management: this is the most contested threshold, as “interventional” in the IVDR context carries a different meaning from “interventional” in clinical trial legislation
- Study design: interventional vs observational
- Sample type: fresh vs leftover, and the level of invasiveness required to collect them
- Risk profile: additional procedures vs standard care
- Whether device endpoints are clearly distinguished: from drug endpoints
Top 12 RFIs From 40+ Combined Programs
This is the data MDCG guidance documents do not provide. Over 40 combined programs, these are the most frequent Requests for Information (RFIs) we have received from NCAs and Ethics Committees across Europe:
1. Insufficient analytical validation (GSPR 9.1a). The test is a clinical trial assay, prototype, or lab-developed test (LDT) and was not developed under full design control. This may be accepted in US early-phase trials but is a common reason for rejection in the EU.
2. Insufficient data supporting the chosen cut-off. A small number of samples were used for cut-off validation. NCAs expect robust, data-driven rationales, not theoretical arguments.
3. Lacking or vague definitions of primary endpoints related to IVD performance, and the absence of a statistical analysis plan for the device component.
4. Informed Consent Form (ICF) lacks performance study-specific content. This is extremely common in combined CT/PS ICFs. Authorities regularly request revision in lay-user language that clearly distinguishes participation in the drug trial from participation in the diagnostic study.
5. Inconsistent objectives between the clinical trial application and performance study documentation. When the CPSP says one thing and the CTA says another, expect an RFI.
6. Incomplete risk plans and reports, with insufficient evaluation in the Investigator’s Brochure of how inaccurate test results could impact the clinical trial, especially for high-toxicity therapies.
7. Investigator and site documentation lacking PS references, required translations, or country-specific insurance certificates.
8. Lack of consideration towards monitoring the IVD study under ISO 20916, especially in sponsor-CRO or multicentre settings.
9. Divergent views between Ethics Committees and NCAs on risk, burden, or benefit, sometimes within the same country.
10. Use of leftover samples for future research without clear documentation on traceability, consent, or ethical approval.
11. Safety monitoring procedures in the CPSP not aligned with IVDR Article 74 and MDCG 2024-4.
12. Scientific inaccuracies in the CPSP, IB, and technical documentation, simple errors that erode credibility and trigger additional scrutiny.
What “Device-Specific Endpoints” Actually Means
One of the most common mistakes we see in combined studies is sponsors relying solely on drug efficacy endpoints (ORR, PFS, OS) as evidence of IVD performance. Under the IVDR and MDCG 2025-5, performance studies must generate data that establish or confirm device performance, not drug performance.
Analytical Endpoints (Often Used in Phase 1/2 Studies)
These include reportable rate, invalid-result rate, repeat-test rate, and assay failure rate; precision and reproducibility in the clinical setting; and accuracy at clinically relevant cut-offs.
Clinical Endpoints (CDx-Type, Often Used in Phase 3 and Bridging)
These include concordance with a clinically valid reference assay (PPA, NPA, OPA); diagnostic sensitivity and specificity vs clinical status; clinical outcomes studies in the IVD-selected group linked to drug endpoints; and clinical bridging studies.
What Is Not Sufficient as a Sole Endpoint
Turnaround time alone, and drug efficacy endpoints (ORR, PFS, OS) without device-specific performance measures.
Case Study: From Rejection to 8-Country Approval
One of the programs presented in detail involved an NGS-based CDx used to detect a specific mutation for targeted therapy patient eligibility in a rare tumour indication. It was a Phase 3 global trial with an interventional combined performance study across 30+ EU sites.
The Initial Rejection
The first EU submission was rejected. The reasons were typical of what we see repeatedly:
- Incomplete analytical performance data vs GSPR 9.1(a) at the mutation cut-off
- Weak linkage between biomarker, clinical condition, and treatment, a scientific validity gap
- Under-developed risk management for false negatives in a high-toxicity therapy
What We Did
MDx CRO conducted a full gap analysis and remediation covering the Scientific Validity Report (SVR), Analytical Performance Report (APR), CPSP, and risk file versus Annex XIII requirements. We rewrote the CPSP with device-specific treatment-decision endpoints, strengthened the analytical data at the decision threshold, and quantified the false-negative impact in the risk management file.
The Outcome
Harmonised resubmission to 8 Member States. All RFIs resolved. Approvals in all 8 countries, within 4 months.
This case illustrates a pattern we see consistently: the initial submission fails not because the science is weak, but because the documentation does not speak the language the NCAs expect. The same data, presented differently, achieves a completely different result.
The Most Costly Mistake Sponsors Make
The single most expensive mistake we have seen sponsors make in the IVDR portion of a combined study is treating the performance study application as an afterthought, something to be “bolted on” after the clinical trial application is already in motion.
In one program, the sponsor had not engaged with IVDR requirements until approximately three months before planned first-patient-in. At that point they discovered that a PSA was required in multiple Member States, that documentation requirements were not harmonised across those countries, and that each required different local forms, different ethics committee interactions, and in some cases certified translations.
The result was a nine-month delay to the European portion of the trial and an estimated additional cost in the range of €800,000-€1.2 million when accounting for CRO renegotiation, site re-engagement fees, amended contracts, and the opportunity cost of delayed clinical data.
This is not an outlier. Industry survey data shows that a significant proportion of sponsors experience 6–12 months of IVDR-related delay in combined programs, with some reporting delays beyond 12 months. The structural cause is always the same: the pharmaceutical team and the diagnostics team are operating on disconnected timelines, and the IVDR submission is not integrated into the master trial timeline from the outset.
Wave Planning: The Operational Playbook
Not all EU Member States are equal when it comes to combined study submissions. One of the practical tools shared at the conference was our wave planning approach, a strategic framework for sequencing country activations based on regulatory architecture, not just commercial priority.
Wave 1: Speed, Parallel/Combined Processes
Spain offers parallel review, with submission to both AEMPS via the portal and the Ethics Committee (usually via email). Total timeline: approximately 85 days.
Belgium uses a consolidated review via CESP where the NCA and EC coordinate and issue a single opinion. Timeline: approximately 60 days.
These are faster because the EC and NCA review simultaneously, not sequentially.
Wave 2: Sequential Requirements
Germany, Austria, and Hungary all require EC approval first, followed by NCA submission. This adds the full EC timeline (46–166 days) before the NCA clock even starts. Total timelines: 135–267 days.
Wave 3: Administrative Complexity
Bulgaria requires notarised and apostilled Powers of Attorney, sworn translations, and physical courier submissions.
Poland requires paper copy submissions to both the EC and NCA, including wet-ink signed site documents, sworn translations, and stricter requirements demonstrating the Sponsor’s business registration and Power of Attorney.
Ireland has non-harmonised reviewers, causing inconsistent RFI rounds.
Why Wave Planning Matters
Typically, it is strategic to avoid submitting to high-administrative-burden countries in Wave 1. For example, Poland necessitates that the entire submission package is delivered to the EC and NCA via courier in printed form with wet-ink signatures. It is more efficient to submit first to countries with accessible pathways so that feedback from RFIs and lessons learned can be incorporated from the start for the more labour-intensive submissions.
The strategy: stagger activation by submission architecture and administrative burden, not just by regulator consolidation.

How MDCG 2025-5 Is Changing the Landscape
MDCG 2025-5, published in June 2025, is the first dedicated Q&A guidance specifically addressing IVDR performance studies comprehensively. It covers 54 questions across topics that sponsors and NCAs have been debating since 2022, including:
- A regulatory pathway decision tree (Appendix I) providing a structured framework for determining whether a planned activity requires a PSA, a PSN, or no submission at all
- Clarification that “interventional” in the IVDR context meaning the results may influence patient management carries a different meaning from “interventional” as used in clinical trial legislation. This conflation has caused significant confusion
- A working definition of “leftover samples” and the conditions under which they trigger notification rather than full application requirements
- Guidance on combined studies including sponsor responsibilities, substantial modification handling across both the CTR and IVDR pathways, and the role of the performance study investigator
Are NCAs Following It?
Partially, and unevenly. Some authorities have begun aligning their pre-submission guidance with the decision-tree logic. Others continue applying their own interpretations, particularly on the definition of “interventional” which remains the most contested threshold.
The guidance is explicitly non-binding: MDCG 2025-5 itself states it cannot be regarded as reflecting the official position of the European Commission and that only the Court of Justice of the EU can give binding interpretations. In practice, guidance adoption across Member States typically lags publication by 12–18 months before a clear majority are operating consistently with it.
The Health Services Pack and the Biotech Act: Will It Actually Simplify Combined Studies?
In December 2025, the European Commission proposed two complementary pieces of legislation as part of a broader health services package. The first is a targeted revision of the MDR and IVDR, aimed at simplifying the existing regulatory framework. The second is the Biotech Act, which proposes amendments to the Clinical Trials Regulation that are directly relevant to combined studies.
What the Biotech Act Changes for Combined Studies
This is the more consequential development. The Biotech Act explicitly introduces a single integrated application for combined studies. Under the current framework, sponsors must seek authorisation of the clinical trial under the CTR and the performance study under the IVDR entirely independently separate portals, separate assessments, separate timelines, separate Member State interactions.
The Biotech Act proposes to eliminate that dual-track requirement. Instead, the sponsor would submit a single application covering both the investigational medicine and the IVD through a combined authorisation process managed under the CTR. That assessment would be led and coordinated by a Reporting Member State (RMS), with coordinated approvals across participating countries.
The proposal also accelerates CTR timelines generally, reducing the multinational CTA review from 106 days to 75 days, and as low as 47 days when no information request is issued.
My Assessment
This is not wishful thinking for the first time, there is a concrete legislative mechanism on the table that directly addresses the structural root cause of combined study delays. If adopted as proposed, it would be a genuine step-change.
However, three cautionary notes:
Legislative timeline. Even with political priority, realistic adoption and implementation timelines are 18–24 months from proposal. Do not expect sponsors to be able to use the single-application pathway before late 2027 at the earliest.
Implementation gap. CTIS will need to be updated to accept performance study documentation alongside the CTA, and Member State competent authorities will need to build assessment capacity for the IVD components within their CTR review teams.
The COMBINE pilot is the bridge. The COMBINE programme’s Project 1 pilot, launched on 13 June 2025, is already testing an “all-in-one” coordinated assessment approach. Sponsors who participate now are effectively rehearsing for the future framework.
The December 2025 package does have the potential to fundamentally simplify combined studies. But sponsors working in 2026 are still operating under the current framework. The practical advice remains: use the COMBINE pilot if you can, plan for Member State divergence if you can’t, and build your combined study submission strategy on the assumption that the current dual-track system will be in place until at least late 2027.
The #1 Mistake Sponsors Make in Their First Combined Study
They assume the IVDR performance study is the diagnostic manufacturer’s problem, not theirs.
The pharmaceutical sponsor designs the trial, defines the biomarker strategy, selects the assay, and writes the protocol. But when the conversation turns to the IVDR submission, they expect their diagnostic partner to handle it independently.
The reality is that under Article 2(57) of the IVDR, the “sponsor” is whichever entity takes responsibility for the initiation, management, and financing of the performance study and in a combined study, particularly in early-phase trials, that is often the pharmaceutical company itself.
This disconnect produces predictable failures:
- The drug application is submitted through CTIS months before the PSA is even drafted
- The diagnostic partner lacks access to the clinical trial protocol, site-level information, and country-specific documentation needed to complete the Annex XIV dossier
- When an NCA issues an RFI at the interface between the two applications, neither team owns the response
MDCG 2022-10 is explicit: the clinical trial sponsor is responsible for overall compliance of products used in the trial, including the IVDR. Where a sponsor uses a CE-marked IVD outside its intended purpose, it assumes manufacturer responsibilities under Article 16(1).
The fix is straightforward: from Day 1 of trial design, the PSA workstream sits inside the integrated trial timeline, with a named owner, shared document management, and joint governance between pharmaceutical and diagnostics teams. Sponsors who do this avoid most problems. Sponsors who don’t are the ones calling us nine months before database lock.
How Much Time Does Expert Guidance Save?
Based on our experience across 40+ combined programs, working with a CRO that has specific IVDR and IVD regulatory expertise saves an average of 3 to 5 months compared to in-house teams or generalist CROs approaching combined studies for the first time.
The savings accumulate in three areas:
Pre-submission strategy (4–6 weeks saved). A specialist knows which Member States to sequence in Wave 1, what each authority actually expects beyond the statutory minimum, and which borderline classification questions need to be resolved before any dossier is submitted. Preventing a single misclassification avoids months of remediation.
Documentation preparation (4–8 weeks saved). Generalist CROs frequently prepare a single PSA dossier and submit it identically across all Member States. A specialist prepares country-adapted packages from the outset correct local forms, country-specific ethics committee requirements, certified translations, and portal-specific submission formats.
RFI management (2–4 weeks saved). When an NCA issues a request for information, a specialist drafts a response that also anticipates how the same question may be raised by other authorities whose reviews are still in progress preventing cascading delays across the wave.
Five Steps to De-Risk Your Next Combined Study
Based on the patterns from 40+ programs, these are the five actions that make the biggest difference:
1. Science first: do not underestimate analytical validation data. Analytical data should be traceable and robust, particularly around the cut-off. The APR and IB should be readable as standalone documents NCAs assess them that way.
2. Decide stakeholder ownership early. Sponsor vs Dx partner vs CRO for both the CTR and IVDR pathways. Ambiguous ownership is the single biggest source of delay.
3. Design device-specific endpoints upfront. Analytical and/or clinical, not just drug efficacy. If your performance study plan only lists ORR as an endpoint, expect a rejection.
4. Build the Annex XIII/XIV package early. SVR, APR, CPSP, risk management, GSPR logic evaluate internal capabilities and gaps before the submission clock starts.
5. Use structured tools for multi-country variation. Don’t rely on memory or email threads. Robust study design combined with an informed country strategy is what separates 60-day approvals from 267-day ones.
Looking Ahead: 2026–2027
From our experience, it will get slightly worse before it gets meaningfully better with the inflection point likely in late 2026 to early 2027.
The short-term pressure comes from volume. More sponsors are now aware of IVDR performance study requirements, which means more submissions entering the system. NCA capacity has not grown proportionally. Several smaller Member States still lack dedicated performance study reviewers.
The medium-term improvement will come from three converging developments: MDCG 2025-5 gradually reducing classification disputes, the COMBINE programme’s coordinated assessment pilot producing procedural lessons by mid-2026, and the December 2025 legislative revision proposal delivering structural changes by late 2027.
The biggest variable remains NCA behaviour. Regulatory guidance adoption is uneven and slow. It typically takes 12–18 months from MDCG publication before a clear majority of authorities are operating consistently with it.
For sponsors planning programs in 2026, the practical reality has not changed: plan for divergence between Member States, budget for country-specific regulatory strategies, engage the IVDR workstream at trial design not after the CTR is submitted and do not assume that what worked in one country will work in the next.
Download the Full Presentation
This article is based on my presentation at the 16th Clinical Biomarkers & Companion Diagnostics Summit Europe in London (March 30, April 1, 2026). The full slide deck includes additional data, the PSA approval timeline map across EU Member States, and detailed case studies that could not be covered in article format.
Need Help With Your Combined Study?
MDx CRO has managed 40+ combined programs across 20+ countries with a 100% approval rate and 90+ performance study submissions in the EU27. Our fastest program went from kick-off to full IVD Study Package Development and submission in 4 weeks.
If you are planning a combined study under the CTR and IVDR, talk to our team about how we can accelerate your path to first patient tested.
Frequently Asked Questions about IVDR Combined Studies
IVDR performance study requirements are triggered through three pathways. Article 58.1 requires authorisation for non-CE-marked devices or devices used outside their intended purpose. Article 58.2 requires notification for companion diagnostics that use leftover samples only. Article 70 requires notification for CE-marked devices that involve additional burdensome procedures. Key decision factors include CE mark status for the specific trial use, whether results affect medical management, the study design, the sample type, and the risk profile.
Approval timelines vary by Member State. The EU COMBINE Analysis Report 2024 shows an average of 137 days, with a range of 45–267 days. Countries with parallel ethics committee and national authority review, such as Spain (~85 days) and Belgium (~60 days), are faster than those with sequential review, such as Germany, Austria, and Hungary (135–267 days).
Based on data from more than 40 combined programs, the most common RFIs include insufficient analytical validation under GSPR 9.1(a), insufficient data supporting the selected cut-off, missing device-specific endpoints in the CPSP, informed consent forms lacking performance study-specific content, and inconsistent objectives between the clinical trial application and performance study documentation.
MDCG 2025-5, published in June 2025, is the first comprehensive Q&A guidance for IVDR performance studies. It introduces a regulatory pathway decision tree, clarifies that “interventional” in the IVDR context differs from the CTR definition, defines “leftover samples,” and addresses sponsor responsibilities in combined studies. Adoption by national authorities is uneven, and full alignment is expected within 12–18 months.
The Biotech Act proposed in December 2025 introduces a single integrated application for combined studies under the CTR framework. If adopted, it would remove the need for dual CTR and IVDR submissions. Realistic implementation is not expected before late 2027. The COMBINE programme pilot is currently testing coordinated assessment as a bridge to the future framework.