Co-authored by Floella Otudeko
The United States is the world’s largest market for in vitro diagnostic devices. It is also one of the most complex to enter. FDA regulation of IVDs operates through a risk-based framework that determines how a device is classified, which approval or clearance pathway applies, what evidence is required before commercial distribution, and what obligations continue after authorization.
This guide covers the entire FDA regulatory landscape for IVD manufacturers, from initial device qualification through classification, premarket submission, investigational use, quality management system requirements, and post-market surveillance. It also addresses the specific considerations that apply to laboratory-developed tests, companion diagnostics, and manufacturers operating in both the US and EU markets simultaneously.
Key point: FDA regulation of IVDs sits within the same statutory framework as all medical devices under the Federal Food, Drug, and Cosmetic Act (FD&C Act). However, IVDs have specific classification rules, dedicated guidances, and performance evidence requirements that differ materially from those applied to other device types.
Align Your Clinical Strategy with FDA IVD Regulations
The upcoming IMDRF N91 framework is designed to align with strict global benchmarks, and the US FDA is leading the charge. Whether you are navigating 510(k) clearances, De Novo pathways, or complex PMA submissions for companion diagnostics (CDx), securing FDA clearance demands impeccable, scientifically valid clinical evidence. To build a bulletproof regulatory strategy that satisfies FDA examiners, you need a partner who understands the nuances of US IVD clinical trial design inside and out.
What Counts as an IVD Under FDA Law
An in vitro diagnostic product is defined under 21 CFR 809.3(a) as a reagent, instrument, or system intended for use in the diagnosis of disease or other conditions, through the in vitro examination of specimens derived from the human body.
The definition is broad. It covers reagent kits, assay systems, instruments that process or read those assays, software that interprets results, and combinations of these elements. A product intended to generate medically actionable information from a human specimen will generally fall within the IVD framework regardless of the format in which it is supplied.
Two practical questions determine whether the FDA IVD regulatory framework applies:
- Is the product intended to examine specimens taken from the human body?
- Is that examination intended to support a medical decision, diagnosis, monitoring, patient selection, or treatment assignment?
If both are true, the product is an IVD and must comply with the applicable FDA requirements before commercial distribution in the United States. The intended use stated by the manufacturer, not the technology, the format, or the distribution channel, determines this status.
IVD vs. LDT: Laboratory-developed tests (LDTs), assays designed, manufactured, and used within a single laboratory, have historically operated under a different framework. FDA issued a final rule in 2024, but subsequent legal challenges (including a 2025 court decision) have created uncertainty, and the long-term regulatory framework for LDTs remains in flux. See the section on LDTs below.
IVD Classification: How FDA Assigns Risk Class
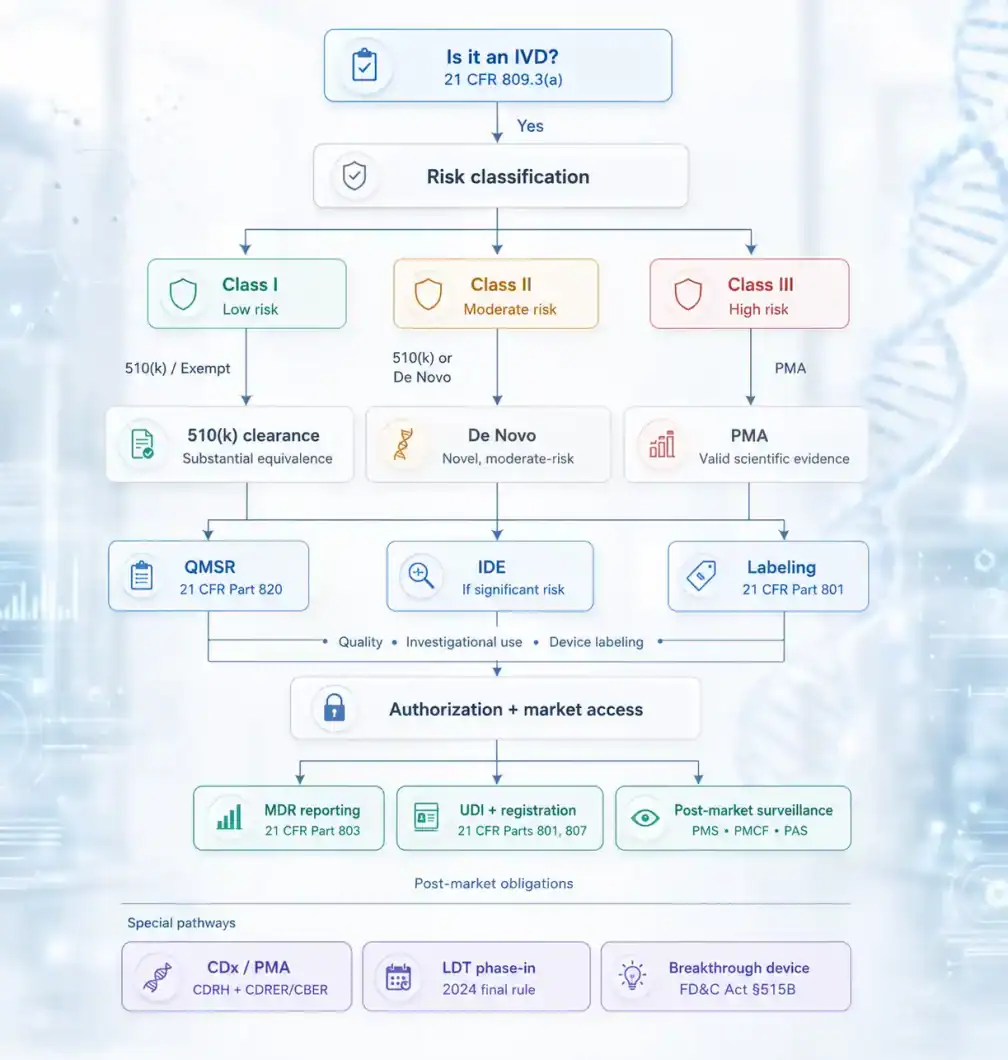
All FDA-regulated devices, including IVDs, are assigned to one of three risk classes. The class determines which controls apply and which premarket pathway is available.
| Class | Risk Level | Controls Required | Typical Premarket Route |
| Class I | Low | General controls only | Exempt or 510(k) |
| Class II | Moderate | General + special controls | 510(k) or De Novo |
| Class III | High | General + special controls + PMA | Premarket Approval (PMA) |
For IVDs, classification is determined primarily by intended use and the clinical consequences of erroneous results, false positives, false negatives, or invalid results, rather than by the underlying technology. An assay with a low-risk intended use (for example, confirming a known condition in a low-risk population) may qualify as Class II even if it uses the same analytical technology as a Class III device with a high-risk intended use.
The FDA maintains a product classification database with assigned device codes, regulation numbers, and review panels. Before finalising a regulatory strategy, manufacturers should search this database for devices with a similar intended use to understand how FDA has previously classified comparable products.
FDA Premarket Pathways for IVDs: 510(k), De Novo, and PMA
The three main premarket pathways differ significantly in what they require, how long they take, and what outcome they produce. Selecting the wrong pathway, or failing to engage FDA early to confirm the right one, is one of the most common and costly mistakes in IVD regulatory strategy.
510(k) Premarket Notification
A 510(k) submission demonstrates that a new device is substantially equivalent to a legally marketed predicate device. For IVDs, this means identifying a predicate with the same intended use and similar technological characteristic, or, if the technology differs, demonstrating that the differences do not raise new safety or effectiveness questions.
The 510(k) route is appropriate when a suitable predicate exists. For IVD manufacturers, substantial equivalence must be established not just at the level of technology but at the level of the clinical claim. An assay using the same analytical platform as a predicate is not automatically equivalent if it is intended for a different patient population, a different decision point, or generates results that would be used differently in clinical practice.
- Standard 510(k): most common; requires performance data and predicate comparison
- Abbreviated 510(k): relies on FDA-recognised performance standards
- Special 510(k): limited to device modifications by the original manufacturer
Timeline: FDA’s standard review target is 90 days. In practice, requests for additional information (AI letters) are common and extend the timeline. Manufacturers should plan for 4–8 months for a standard 510(k) with a well-prepared submission.
De Novo Risk-Based Classification for Novel Devices
The De Novo pathway applies to novel devices for which no legally marketed predicate exists, making the 510(k) pathway unsuitable, but the device is not high enough risk to require PMA. It creates a new device classification with specific controls appropriate to that device type.
For IVD manufacturers developing novel diagnostic technologies or applying known assay types to new clinical applications, De Novo is often the appropriate route. A successful De Novo classification becomes itself a predicate for future 510(k) submissions by other manufacturers.
The evidentiary requirements for De Novo are substantial and in practice often comparable to those for a PMA in terms of analytical and clinical evidence depth. The key difference is that De Novo does not presuppose Class III and gives FDA the ability to define special controls that make Class II regulation workable for the device type.
Timeline: FDA’s target is 150 days for De Novo review. With AI letters, 12–18 months is a realistic planning assumption for novel IVD applications.
PMA Premarket Approval
Premarket Approval is the most rigorous FDA pathway and applies to Class III devices, those for which general and special controls are insufficient to assure safety and effectiveness. PMA requires valid scientific evidence, typically from well-controlled clinical investigations, demonstrating that the device is safe and effective for its intended use.
For IVDs, PMA applies to:
- Devices that determine eligibility for high-risk therapies where no predicate or De Novo classification exists
- Companion diagnostics essential for the safe and effective use of a corresponding therapeutic, the majority of CDx devices proceed through PMA, though a November 2025 FDA proposal would reclassify certain NGS and NAAT-based oncology CDx to Class II
- High-risk screening tests where a false negative or false positive result has severe clinical consequences
PMA submissions are organised in modules covering administrative information, device description, clinical performance data, analytical validation, manufacturing, software, and labeling. FDA offers a modular PMA submission option that allows review of completed modules before the full package is assembled, a significant strategic advantage in co-development programmes where evidence is generated progressively.
| Criterion | 510(k) | De Novo | PMA |
|---|---|---|---|
| Legal standard | Substantial equivalence | Safety & effectiveness via general/special controls | Safety & effectiveness |
| Predicate required | Yes | No | No |
| Device class outcome | Class I or II | New Class I or II | Class III |
| Review target | 90 days | 150 days | 180 days (modular: staged) |
| Post-approval changes | Change assessment required | Change assessment required | PMA supplement required |
| Typical IVD use case | Known assay type, clear predicate | Novel technology, moderate risk | CDx, high-risk screening, no predicate |
Investigational Use of IVDs: IDE Requirements and the Significant Risk Determination
When an IVD is used in a clinical investigation before it has received FDA premarket authorisation, the manufacturer and sponsor must comply with the investigational device provisions under 21 CFR Part 812. The applicable requirements depend on whether the device is classified as significant risk (SR) or non-significant risk (NSR).
Significant Risk vs. Non-Significant Risk
A significant risk device is one that presents a potential for serious risk to the health, safety, or welfare of a subject, including devices used in diagnosing, curing, mitigating, or treating disease where errors could have serious clinical consequences.
Investigational IVDs Used in Clinical Investigations of Therapeutic Products
For IVDs, the SR determination requires analysis of four specific questions defined in FDA’s 2017 draft guidance on Investigational IVDs Used in Clinical Investigations of Therapeutic Products:
- Will use of the IVD results lead some subjects to forgo or delay a treatment known to be effective?
- Will use of the results expose subjects to safety risks exceeding those of the control arm or standard of care?
- Is it likely that incorrect results would present a potential for serious risk, based on what is known about the biomarker-therapeutic relationship?
- Does use of the IVD require invasive sampling that is not part of standard care?
Sponsors are responsible for the initial risk determination and must present it to the Institutional Review Board (IRB). If the device is classified as SR, an Investigational Device Exemption (IDE) application must be submitted to FDA (through CDRH) and FDA has 30 days to respond. If deemed NSR, IRB approval is sufficient, no IDE application is required, but abbreviated requirements under 21 CFR 812.2(b) still apply.
CDx in drug trials: When an IVD is used in a clinical investigation of a drug or biological product to determine patient eligibility or guide treatment decisions, the SR determination is critical. A misjudgement here, treating a device as NSR when it is SR, exposes the entire clinical programme to regulatory risk.
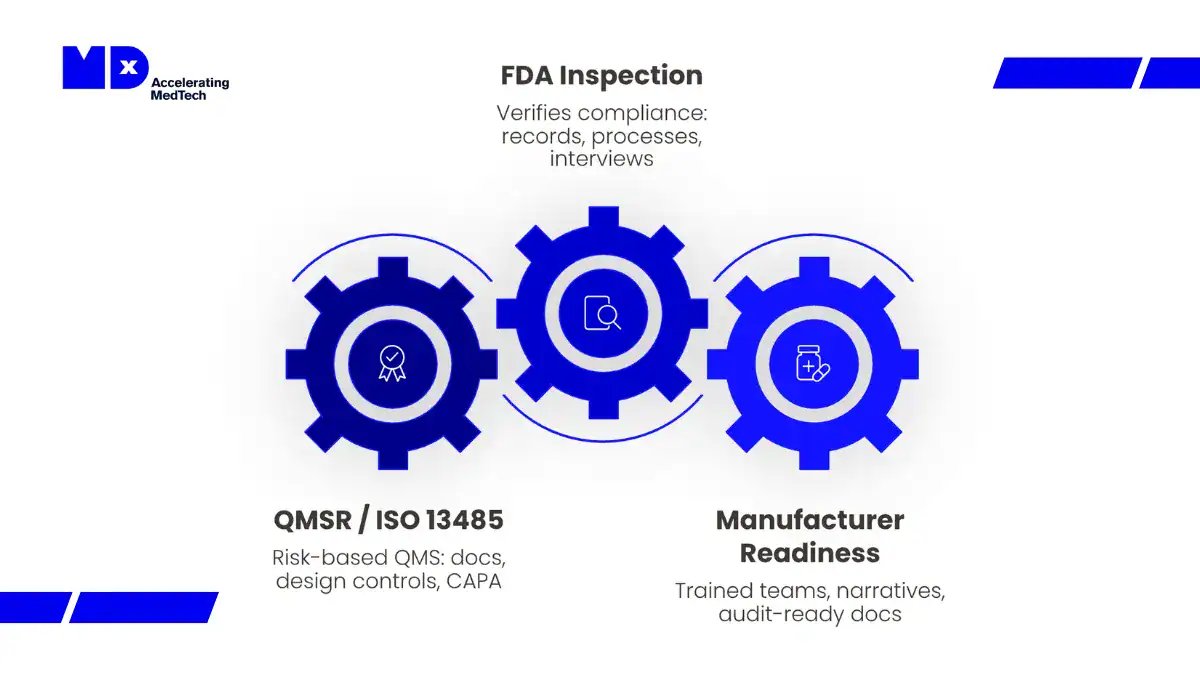
FDA Quality Management System Regulation (QMSR): What IVD Manufacturers Need to Know
Since 2 February 2026, the FDA Quality Management System Regulation (QMSR, 21 CFR Part 820) has replaced the former Quality System Regulation (QSR). QMSR incorporates ISO 13485:2016 by reference into US law, aligning US quality system requirements with the international standard for the first time.
For IVD manufacturers, QMSR introduces important practical changes. Several are particularly relevant to the IVD context:
What Changed for IVD Manufacturers Specifically
IVD quality systems under QMSR must go further than a standard ISO 13485-aligned QMS. The nature of IVD products, which generate results that directly inform clinical decisions, introduces additional complexity in several areas:
- Design controls (ISO 13485 clause 7.3 / QMSR): design inputs must explicitly link intended use, specimen type, analyte definition, and performance claims to statistically robust analytical and clinical data. Design changes after clinical evidence generation may require bridging studies.
- Risk management: for IVDs, risk management must account for false positive and false negative results, not just device failures. Diagnostic decision risks, interfering substances, and matrix effects must all be explicitly addressed.
- Production and process control: lot-to-lot variability of biological materials requires validation against clinically relevant criteria, not just functional performance.
- Post-market surveillance: systems must detect performance drift, shifts in sensitivity or specificity, which may not be visible through standard complaint handling alone.
The ISO 13485 Certification Gap
A persistent misconception in the IVD industry is that ISO 13485 certification means a manufacturer is QMSR-ready. It does not. While QMSR incorporates the standard by reference, FDA-specific requirements remain fully enforceable, and ISO certification does not address them.
The most consistently overlooked gaps include:
- UDI traceability: the UDI must be recorded consistently across complaints, servicing records, and batch/history records, not just registered in GUDID
- MDR linkage: complaint handling procedures must be explicitly connected to Medical Device Reporting obligations under 21 CFR Part 803
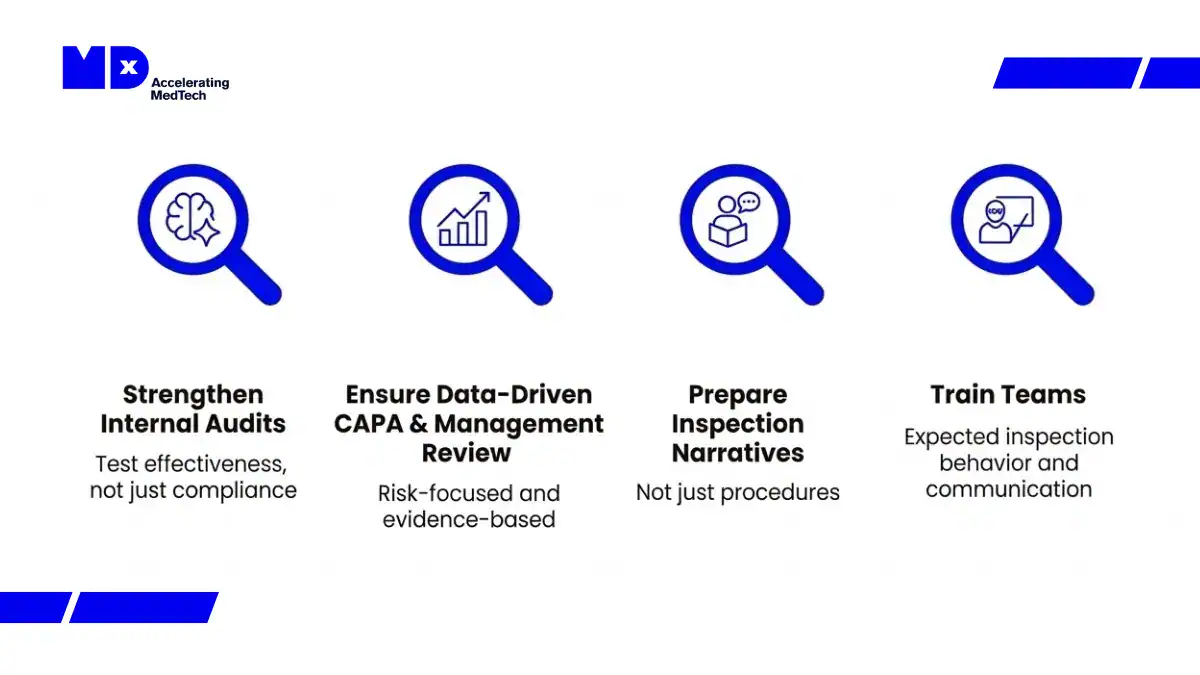
- Internal audit and management review access: under QMSR, FDA inspectors can access these records, they were previously outside standard inspection scope
- Labeling controls: FDA-specific label content requirements (UDI, expiry dates, handling instructions) require documented control procedures
Inspection change: Under QMSR and Compliance Program 7382.850, FDA inspections are now structured around six integrated quality system areas rather than the former QSIT subsystem model. Inspectors assess how quality processes function together across the product lifecycle, not whether each subsystem satisfies a checklist in isolation.
For manufacturers needing support with QMSR gap assessment, MDx CRO’s quality and regulatory affairs team provides structured readiness reviews specifically for IVD and CDx manufacturers. See the dedicated article: FDA QMSR: How the 2026 Regulation Shift Transforms MDSAP Audits and FDA Compliance Inspections.
Laboratory-Developed Tests (LDTs): FDA’s 2024 Rule and What It Means
FDA issued a final LDT rule in May 2024 that would have phased out enforcement discretion. However, the rule was vacated by a federal court in March 2025, and FDA later reverted the regulatory text. The future federal regulatory framework for LDTs remains uncertain.
| Phase | Timeline (from May 2024) | Requirement |
|---|---|---|
| 1 | Year 1 | MDR, complaint handling, correction/removal reporting |
| 2 | Year 2 | Registration, listing, labeling, investigational use |
| 3 | Year 3 | Quality system (QMSR) requirements |
| 4 | Year 4 | Premarket review — Class III LDTs |
| 5 | Year 5 | Premarket review — Class II LDTs (most) |
The practical implication for laboratories offering LDTs is significant. Tests that have operated without FDA premarket review for decades now face classification, potential 510(k) or PMA requirements, QMSR compliance, and post-market surveillance obligations. The transition is particularly complex for high-complexity molecular tests, NGS-based assays, and tests used to guide treatment decisions, the categories most likely to face substantive premarket requirements.
The IVD vs. LDT distinction has also created regulatory ambiguity for assays that are commercially distributed but described as ‘laboratory services’. FDA’s position is that the legal framework applies based on the nature of the product and its intended use, not the business model through which it is offered.
Companion Diagnostics: FDA’s Co-Development and Approval Framework
A companion diagnostic is an IVD that provides information essential for the safe and effective use of a corresponding therapeutic product. FDA’s CDx framework sits at the intersection of device and drug regulation, requiring coordination between CDRH (which reviews the diagnostic) and CDER or CBER (which reviews the drug or biologic).
Most companion diagnostics require PMA, though FDA’s November 2025 proposal to reclassify certain NGS and NAAT-based oncology CDx from Class III to Class II would, if finalised, make 510(k) available for a subset of these devices. Until that reclassification is confirmed, PMA should be treated as the working assumption for novel CDx programmes.
The Contemporaneous Approval Expectation
FDA’s CDx policy generally expects the companion diagnostic and the corresponding therapeutic product to receive marketing authorization simultaneously. This means that a deficiency in the diagnostic submission, a gap in analytical validation, an incomplete bridging study, or an unresolved labeling alignment issue, can delay drug approval, even though the drug is reviewed by a different FDA centre.
The most common causes of CDx-related drug approval delays are:
- Assay version used in the clinical trial differs from the commercial assay proposed for marketing, requiring bridging studies that were not planned in advance
- Clinical performance data collected in one population (for example, EU samples) not adequately bridged to the US population the labeling claim would cover
- Intended use statement in the device submission misaligned with the indication in the drug label
- Design controls documentation not adequately tracing the clinical trial assay to the commercial configuration
MDx CRO’s FDA Companion Diagnostics guide covers the full CDx regulatory pathway including PMA structure, IDE strategy, Q-submission programme, co-development governance, and assay lock considerations. A downloadable FDA Companion Diagnostic Roadmap is available on that page.
FDA Regulation vs. EU IVDR: Key Differences for IVD Manufacturers Targeting Both Markets
Many IVD manufacturers pursue US FDA authorisation and EU CE marking under the IVDR simultaneously. While the two frameworks share some structural principles, risk-based classification, performance evidence requirements, quality management system obligations, they diverge significantly in how those principles are applied.
| Area | FDA (US) | EU IVDR |
|---|---|---|
| Classification basis | Intended use + clinical risk | Risk class A–D based on IVDR rules + MDCG guidance |
| Premarket route | 510(k), De Novo, or PMA | Conformity assessment via Notified Body (Class B–D) or Self-certication (Class A) |
| Evidence standard | Valid scientific evidence (PMA) / substantial equivalence (510(k)) | Scientific validity + analytical + clinical performance |
| Clinical evidence location | US population; bridging required for non-US data | EU population; bridging required for non-EU data |
| QMS standard | QMSR (incorporates ISO 13485:2016) | ISO 13485:2016 (harmonized under the IVDR) |
| Performance studies | IDE framework (21 CFR Part 812) | IVDR Articles 56-58 / Annex XIV |
| Post-market | MDR reporting, post-approval studies (PMA) | PSUR, PMPF, vigilance reporting |
| CDx specifics | CDRH/CDER/CBER coordination; PMA typical | Notified Body review; Annex IX section 5.2 procedure |
The most significant practical challenge for dual-jurisdiction programmes is clinical evidence. A clinical performance study conducted exclusively with EU samples may not be acceptable to FDA without bridging evidence demonstrating equivalence to the US population. The reverse applies equally. Early engagement with both FDA (via Q-submission) and the Notified Body allows sponsors to design studies that generate evidence acceptable to both frameworks, reducing the need for duplicated studies.
Analytical validation can often be leveraged across jurisdictions if conducted to CLSI and CAP/CLIA standards, which both frameworks recognise. Intended use statements can be aligned between jurisdictions, though the labeling requirements differ. The most efficient approach is to map the two frameworks at programme inception and identify shared requirements before study design is finalised.
How MDx CRO Supports IVD Manufacturers Targeting the US Market
MDx CRO is a specialist contract research and regulatory organisation focused on IVDs, companion diagnostics, and medical devices. Our regulatory affairs and quality teams support IVD manufacturers entering or expanding in the US market across the full regulatory lifecycle.
Regulatory Strategy and Pathway Determination
We help manufacturers determine the appropriate FDA classification, premarket pathway, and investigational use strategy before resources are committed. For manufacturers also pursuing EU IVDR CE marking, we map the two frameworks in parallel to identify shared and divergent requirements early.
QMSR Gap Assessment
We conduct structured QMSR readiness assessments for IVD and CDx manufacturers, identifying gaps between current quality systems and FDA QMSR requirements, prioritising findings by inspection risk, and supporting remediation. Our quality team has specific IVD expertise, covering the additional complexity that performance claims, clinical evidence traceability, and analytical validation introduce into design controls and risk management under QMSR.
IDE and Investigational Use Support
We support the significant risk determination process, IDE application preparation, and coordination between the IVD and drug sponsors in combined studies. This includes IVDR performance study applications for EU clinical sites running in parallel with FDA IDE submissions for US sites.
Companion Diagnostic Co-Development
For pharma sponsors co-developing an IVD with their therapeutic programme, we provide IVD regulatory strategy, assay lock planning, bridging study design, and submission preparation, with the goal of achieving contemporaneous FDA authorization of the CDx alongside the drug or biologic.
Frequently Asked Questions
A commercial IVD is a product manufactured and distributed to multiple laboratories or end users. An LDT is an assay designed, manufactured, and used within a single CLIA-certified laboratory. Historically FDA did not enforce premarket requirements against LDTs, but its 2024 final rule phases in the same regulatory framework for LDTs as for commercial IVDs over a four-year period beginning May 2024.
No. 510(k) results in FDA clearance, not approval. Clearance confirms substantial equivalence to a predicate device. FDA approval, issued through PMA, applies to Class III devices and requires independent demonstration of safety and effectiveness. The distinction matters clinically and commercially: the evidentiary standard and post-market obligations differ significantly.
It can be submitted, but FDA will assess whether the evidence is adequate for the US intended use and patient population. For devices where clinical validity depends on population-specific biomarker prevalence, disease incidence, or treatment patterns, bridging evidence may be required to establish that EU data is applicable to the US population. This should be discussed with FDA via Q-submission before the clinical study is designed.
When an IVD is used in a clinical investigation of a drug or biological product and classified as a significant risk device, the sponsor must submit an IDE application to FDA under 21 CFR Part 812. FDA has 30 days to approve or disapprove the application. If the device is non-significant risk, only IRB approval is required. The risk determination must be made by the sponsor and presented to the IRB before the study begins.
ISO 13485 certification does not equal QMSR compliance. While QMSR incorporates ISO 13485:2016 by reference, FDA-specific requirements remain fully enforceable, including UDI integration across record types, MDR linkage to complaint handling, and specific labeling control documentation. Additionally, under QMSR, FDA inspectors can now access internal audit reports, management reviews, and supplier audits, which were previously outside inspection scope. A gap assessment against QMSR requirements is recommended even for certified manufacturers.
FDA’s policy for companion diagnostics that are essential for the safe and effective use of a therapeutic product generally expects simultaneous authorization of both products. A device deficiency can delay drug approval even when the therapeutic programme itself is ready. This makes early integration of the CDx regulatory programme into the drug development timeline, and coordination between CDRH and CDER or CBER, a practical necessity rather than an optional best practice.
The FDA IVD Regulatory Framework at a Glance
FDA regulation of IVDs operates through a risk-based classification system that assigns devices to three classes and directs them toward 510(k), De Novo, or PMA premarket pathways depending on risk level and the availability of a predicate. Investigational use of IVDs in clinical studies requires an IDE for significant risk devices. Commercial distribution requires compliance with QMSR quality management system requirements, which have been in force since February 2026 and now give FDA investigators access to records previously outside inspection scope.
Laboratory-developed tests are progressively subject to the same framework under FDA’s 2024 rule. Companion diagnostics follow the general device framework but require coordination with the drug regulatory review and are generally subject to PMA. Manufacturers pursuing both US FDA authorization and EU IVDR CE marking can leverage shared analytical validation data but must address population-specific clinical evidence requirements for each jurisdiction.