This IVDR Annex XIV clinical performance study guide explains how you can plan and obtain authorisation for performance studies under Annex XIV of the IVDR when it involves a companion diagnostic. It aims to be practical and aligned with current expectations of ethics committees and competent authorities in the European Union.
If you require a structured checklist, you can download the Annex XIV Performance Study Authorisation (PSA) Toolkit, including an ISO 20916 monitoring checklist, templates, and a pre-submission workplan.
Cleared for Enrollment: A Practical Guide to IVDR Annex XIV Studies
Download the white paper here.
IVDR Annex XIV Clinical Performance Study: When PSA vs PSN Applies for Companion Diagnostics
Companion diagnostics (CDx) often require an IVDR Annex XIV clinical performance study because these tests directly influence patient management. Therefore, understanding when a Performance Study Authorisation (PSA) or a Performance Study Notification (PSN) applies is critical for effective regulatory planning and avoiding unnecessary delays.
Why Companion Diagnostics Often Fall Under Annex XIV
Companion diagnostics frequently fall under Annex XIV of the IVDR because the test result guides key treatment decisions, including:
- Patient selection
- Treatment allocation
- Therapy continuation or discontinuation
If the study design allows test results to influence clinical decisions, regulators consider the study interventional, and this classification triggers the need for a Performance Study Authorisation (PSA). In addition, if you use the device outside its intended purpose as defined in the Instructions for Use (IFU), the IVDR framework also requires a PSA.
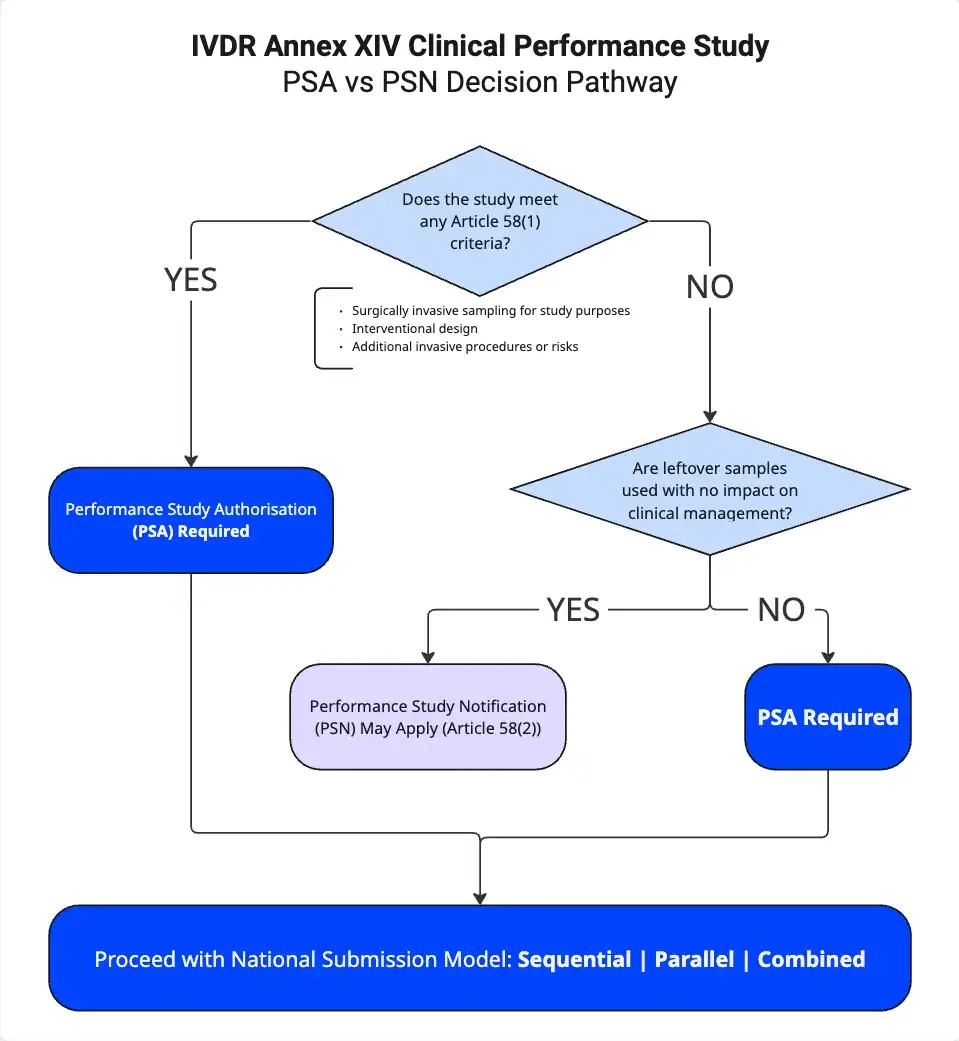
When Does a PSA Apply Under Article 58(1)?
For any IVDR Annex XIV clinical performance study, Article 58(1) serves as the key provision to determine whether a PSA is required. A PSA becomes mandatory if you meet any of the following three criteria:
- You perform surgically invasive sample collection specifically for the clinical performance study (CPS).
- You design the study as interventional in nature.
- You introduce additional invasive procedures or other risks for participants.
If even one of these criteria applies, you must obtain a Performance Study Authorisation before starting the study.
When Does a PSN Apply Under Article 58(2)?
If you do not meet any of the Article 58(1) criteria, Article 58(2) may apply instead. In that case, you may submit a Performance Study Notification (PSN) when:
- The study uses leftover samples only
- The study includes no additional invasive procedures
- Test results do not influence patient management
- The design remains strictly non-interventional
However, even when these conditions apply, you must carefully evaluate national requirements and specific study design details to confirm that a PSN remains appropriate.
Combined Medicinal Product and Diagnostic Studies
An IVDR Annex XIV clinical performance study that involves both a companion diagnostic and a medicinal product requires structured coordination from the outset. When both regulatory frameworks apply, the Clinical Trials Regulation (CTR) governs the medicinal product, while the IVDR governs the diagnostic. Consequently, you must align timelines, documentation, and regulatory strategy under both frameworks to avoid inconsistencies and delays.
Step-by-Step: from planning to PSA approval
1. IB and CPSP Essentials, and the Link Between Endpoints, Intended Use, and Cut-off Strategy
Begin with a coherent Investigator’s Brochure (IB) and Clinical Performance Study Plan (CPSP). Every claim in the CPSP should trace to the intended purpose of the device and to analytical and clinical evidence that is sufficient for that claim. Endpoints must align with the intended clinical decision.
Based on our experience with more than 100 projects, the following two review findings occur repeatedly:
- Analytical cut-off and validation. Authorities closely examine how the assay cut-off has been defined and supported. Sensitivity, precision, and accuracy should demonstrate that the device performs in a way that supports safe clinical decisions. Weak justification invites questions about patient misclassification risk, which frequently leads to requests for information.
- Endpoints not aligned with intended use. Reviewers frequently question primary endpoints that are not clearly tied to clinical performance or to the intended use of the device. The endpoint should map directly to the decision being made for the patient.
Practical measures we have implemented in more than 100 projects:
- Draft the statistical analysis plan early and show a clear line from intended use to endpoint to success criteria.
- Map each analytical study (limit of detection, limit of quantitation, precision, interference, cut-off justification) to the clinical claim it supports.
2. Country Submissions: ethics committees, competent authorities, portals, translations, and fees
Plan both the ethics and competent authority pathways, including accounts for national portals, translation policies, and fee payments. Country-specific requirements can change timelines and logistics.
Some examples of country-specific requirements:
- France requires an IDRCB registration code. The protocol, informed consent form, and insurance certificate must display this code. Missing or inconsistent use of the code commonly triggers requests for information.
- Poland may require a physical submission package rather than a fully electronic file. Plan accordingly all documents that require original signatures. Courier time, notarised copies where applicable, and signature sequencing should also be built into the schedule.
A brief pre-submission checklist:
- Identification of country EC-NCA submission approach (sequential, parallel, combined) and EC meeting schedules
- Portal access verified and roles assigned for both ethics committees and competent authorities.
- National identifiers obtained and propagated consistently across documents.
- Translation scope defined and quality-controlled, particularly for patient-facing materials.
- Insurance certificates aligned with the study footprint and national expectations.
- Fee tables confirmed and purchase orders in place.
3. Timelines, Clock-Stops, and Expert Consultations
Validation and assessment phases of review often include clock-stops for clarification, where the reviewing authority can ask for further information, known as Request for Informations (RFIs). You should define internal service levels for responses in advance, and topic ownership should be clear across analytical, clinical, and biostatistics contributors. A master cross-reference that links CPSP, IB, risk management, and statistical sections reduces the risk of inconsistent responses. In Pickett’s assessment, assigning topic ownership for analytical, clinical, and statistical responses before submission helps keep clock-stops short and prevents inconsistent answers across documents.
IVDR Annex XIV Clinical Performance Study: How to Build a Robust Dossier?
A strong IVDR Annex XIV clinical performance study dossier reduces the risk of Requests for Information (RFIs), clock-stops, and approval delays.
Analytical Validation and Cut-Off Justification
For an IVDR Annex XIV clinical performance study, cut-off justification must go beyond presenting a single threshold value or ROC curve.
A robust dossier should:
- Explain the clinical consequences of the selected cut-off
- Describe how sensitivity and specificity change if the threshold shifts
- Address false positives and false negatives at clinically relevant prevalence
- Link analytical performance directly to the primary endpoint
- Demonstrate how the device supports a safe clinical decision
Authorities frequently focus on misclassification risk. If the cut-off rationale does not clearly support safe decision-making, this section becomes a major driver of RFIs.
Best practice according to Callum Pickett Clinical Alliance Lead at MDx
Treat cut-off justification with the same rigor as a safety argument. Provide both statistical evidence and a clear clinical narrative.
Informed Consent Strategy Aligned With the CPSP
Misalignment between the Clinical Performance Study Plan (CPSP) and the informed consent form is a common cause of delay in an IVDR Annex XIV clinical performance study.
To reduce risk:
- Finalize the CPSP first.
- Draft the informed consent to mirror procedures, visit schedules, and risks.
- Use clear, plain language that accurately reflects the protocol.
Reviewers assess whether participants are properly informed. If the consent document does not reflect the study design, an RFI is likely.
We recommend to include the following in the Informed Consent:
- Clear summary of procedures and assessment schedule
- Device-specific risks, including sample handling and possible retesting
- Explanation of invalid or indeterminate results and participant implications
Consistency between the CPSP and consent documentation strengthens credibility during assessment.
Cross-Referencing: CPSP, IB, GSPR, and Study Reports
A cross-reference matrix improves both internal quality control and external review efficiency.
Your matrix should demonstrate:
- Where each General Safety and Performance Requirement (GSPR) is addressed
- How CPSP procedures are monitored and documented
- Where statistical commitments are supported by analysis
- How risk management links to study controls
For a successful IVDR Annex XIV clinical performance study submission, document traceability is critical.
Frequent RFI Drivers in IVDR Annex XIV Clinical Performance Studies
Below are common deficiencies and practical mitigation strategies:
| RFI Driver | How to Address It |
|---|---|
| Primary endpoint not aligned with intended use | Redefine or restate the endpoint so it directly supports the clinical decision |
| Cut-off justification insufficient | Provide complete analytical data and explain clinical impact |
| Sample representativeness unclear | Justify matrix type, disease stage, prior therapy, and relevant variables |
| Misclassification of study type (leftover samples) | Clarify whether the design remains non-interventional and whether PSN is appropriate |
| Monitoring plan not aligned with ISO 20916 | Define adverse event categories, roles, and timelines |
| Informed consent inconsistent with CPSP | Align language and procedural details precisely |
| Statistical assumptions not clinically justified | Link alpha and power to meaningful clinical differences |
| Device deficiency reporting unclear | Define detection, escalation, and reporting mechanisms |
| Risk management not connected to study controls | Trace risks to mitigation and monitoring activities |
| Combined CTR–IVDR governance unclear | Define roles, responsibilities, and decision pathways |
| Incomplete or low-quality translations | Plan professional review and back-translation |
| National identifiers or insurance mismatched | Ensure consistent codes and appropriate coverage limits |
Country Playbook: How to plan Performance Study Applications to EU Member States
1. Identify the PSA submission approach adopted by the EU member state.
There are three models adopted by EU countries which will impact your submission strategy:
- Sequential – the EC is submitted to first and NCA submission can only occur once an EC approval has been issued.
- Parallel – the EC and NCA submissions can be submitted around or at the same time allowing for a “parallel” review process. However, the NCA will only approve the study once a positive EC opinion has been granted
- Combined – a single PSA submission is sent to one authority which serves as the EC and NCA, a single positive opinion will be issued.
2. Choose your Ethics Committee:
- It is highly recommended to submit the PSA to the same EC reviewing the associated Clinical Trial Application
- Identify any EC specific templates, this may include EC-specific application forms and site document templates
- Identify the EC meeting schedule and the deadlines for PSA submission to achieve review at the EC meeting date
- Use the EC meeting schedule to inform your submission strategy, different ECs will meet at different frequencies.
3. Identify any specific requirements set by the National Competent Authority:
- Are there any NCA specific templates to be filed with the PSA?
- Is there anything that can gate submission to this country? For example, does the NCA mandate that the final Clinical Trial protocol is submitted with the submission.
4. Identify any specific laws and requirements for the EU member state being submitted to:
- The EU is governed by GDPR laws, but national laws on data protection add an additional layer of requirements. Ensure that your study is developed with the national data protection requirements in mind.
- EU member states have different requirements for the insurance documentation, this may include reference to national laws, inclusion of national study codes, and extra details on the number of participants.
5. Use all these points to create your submission strategy, informed by the following:
- Submission approach: countries with sequential review approaches take longer on average than countries with parallel and combined review approaches.
- Clinical Priority: what countries are priority for enrolment? Which countries will have the most sites and therefore need to be activated earlier?
- How often do the chosen ECs meet according to their meeting schedule?
- Are there any NCA or EC document requirements which aren’t yet available, and might delay submission?
Monitoring in Line with ISO 20916
ISO 20916 introduces additional classification categories for adverse events compared with the base IVDR text. Sites need clear training on event taxonomy, responsibilities for classification, and reporting timelines.
Content to include in the monitoring plan and site training:
- Definitions and examples for adverse events and serious adverse events as used in the study.
- Roles for initial classification, medical review, and final assessment.
- Specific clocks for reporting from site to sponsor and from sponsor to authorities.
- How to capture assess and report device deficiencies.
As our clinical team has observed under Annex XIV submissions, early training on adverse event taxonomy and reporting timelines is essential. Misclassification in the first reported case often leads to corrective actions and schedule impact.
Scientific Validity within Annex XIV Performance Evaluation
From PSA to Market: coordination with the notified body and medicines regulators
For a true companion diagnostic approval, align analytical validity, clinical performance, and scientific validity with post-market plans. Label language and evidence expectations should be coordinated with the Notified Body and, where applicable, medicines regulators. Plan the handover from study evidence to post-market performance follow-up.
According to Callum Pickett, maintaining a single evidence map that links analytical validity, clinical performance, and scientific validity to the eventual label language streamlines Notified Body review and reduces post-submission clarification rounds.
Resources
- Guidance from the Medical Device Coordination Group (MDCG) and the European Commission on performance studies, including Q&A on Article 58 pathways.
- National guidance such as the Belgian Federal Agency for Medicines and Health Products (FAMHP) on dossier structure, timelines, and fees for performance studies.
- Consultancy overviews such as DLRC Group (DLRC Group) for pan-EU context.
- Standards published by the International Organization for Standardization (ISO), notably ISO 20916 for clinical performance studies of in vitro diagnostic medical devices.
Cleared for Enrollment: A Practical Guide to IVDR Annex XIV Studies
Download the white paper here.
Expert insight by Callum Pickett
Success with Annex XIV studies for companion diagnostics depends on alignment. Intended use, endpoints, analytical validation and cut-off, consent, and monitoring must be consistent and mutually supportive. Careful attention to country-specific requirements and early planning for CTR-IVDR coordination reduces the likelihood of clock-stops and requests for information. A structured checklist and disciplined cross-referencing improve dossier quality and assessment efficiency.
Read more about IVD clinical studies services.
Frequently Asked Questions (FAQ)
A PSA is required under Article 58(1) if the study includes surgically invasive sample collection specifically for the study, uses an interventional design where test results influence patient management, or introduces additional invasive procedures or risks. In practice, companion diagnostics often trigger a PSA because their results guide treatment decisions. Therefore, as soon as one of these criteria applies, you must obtain a PSA before starting the study. For broader context on running studies under IVDR, read the following article on Running Clinical Studies Under IVDR
You can use a PSN under Article 58(2) when the study remains strictly non-interventional. For example, the study may rely only on leftover samples, avoid additional invasive procedures, and ensure that test results do not influence clinical decisions. However, you must assess the design carefully, because misclassifying a study as non-interventional frequently leads to delays and reclassification requests.
In combined studies, you must comply with both frameworks simultaneously: the CTR governs the medicinal product, while the IVDR governs the companion diagnostic. As a result, you should align endpoints, intended use, and patient population across both submissions from the outset. Otherwise, inconsistencies between the CTR and IVDR dossiers often trigger Requests for Information and clock-stops. A structured gap analysis can help you identify and resolve these risks early. Read the article about pre submission assessment here.
Authorities typically issue RFIs when sponsors fail to align primary endpoints with the intended use, provide insufficient analytical validation or cut-off justification, or clearly explain misclassification risk. In addition, inconsistencies between the CPSP and informed consent, or weak traceability between risk management and statistical assumptions, often raise concerns. Therefore, you should build a clear cross-reference structure across all documents to reduce review friction.
For more detail on documentation expectations, read IVD technical documentation.
To reduce timelines, you should define endpoints early and link them directly to intended use, justify analytical cut-offs with both statistical evidence and clinical rationale, and align the informed consent precisely with the CPSP. At the same time, confirm national submission models, ethics committee schedules, translation scope, and insurance requirements before submission. By assigning clear internal ownership for analytical, clinical, and statistical responses, you can also shorten clock-stops and maintain consistency during review.