Guía para CRO sobre la validación analítica de DIV: Mejores prácticas y errores comunes
septiembre 28, 2025
Dominar la validación analítica de DIV según el IVDR
La validación analítica es la piedra angular del desarrollo de diagnósticos in vitro (DIV). Según el Reglamento de la Unión Europea sobre Diagnósticos In Vitro (IVDR; EU 2017/746), proporciona evidencia de que un dispositivo DIV funciona según lo previsto: de forma precisa, fiable y consistente dentro de su alcance definido. Tanto para los fabricantes con marcado CE como para los laboratorios que operan según el Artículo 5(5), la validación analítica se sitúa en el centro del marco de evaluación del rendimiento en el Artículo 56 y el Anexo XIII.
Parámetros centrales del rendimiento analítico
El IVDR exige una demostración completa de las características de rendimiento analítico en el Anexo I, Sección 9.1(a). Estas incluyen:
Veracidad (sesgo): Cercanía de la concordancia entre los valores medidos y una referencia (ISO 5725-1; JCGM 200:2012).
Precisión: Repetibilidad y reproducibilidad entre instrumentos, operadores y tiempo (CLSI EP05-A3; ISO 20776-2).
Exactitud: Combinación de veracidad y precisión; esencial para obtener resultados fiables.
Sensibilidad analítica (límite de detección, LoD): Cantidad más pequeña de analito distinguible del fondo (CLSI EP17; MM06).
Especificidad analítica: Capacidad para medir solo el analito objetivo, evitando la reactividad cruzada y la interferencia (ISO 15193; CLSI MM09, MM26).
Linealidad: Respuesta proporcional en un rango de concentración definido (CLSI EP06, EP10).
Límite de cuantificación (LoQ): Concentraciones mínimas (y, cuando corresponda, máximas) cuantificables con un error aceptable.
Valores límite: Umbrales que separan los resultados positivos de los negativos (CLSI EP12, EP24).
Rango informable: Intervalo de valores que el ensayo puede notificar de forma fiable.
Trazabilidad metrológica: Vincular los resultados a las referencias a través de una cadena de calibración documentada (JCGM 200:2012).
Estabilidad: Vida útil y estabilidad en uso (ISO 18113-1; EN ISO 23640).
Tipo de muestra y estabilidad: Valide todos los tipos de muestra y condiciones de almacenamiento relevantes (CLSI M47).
Si un parámetro no se aplica, proporcione una justificación sólida. Los reguladores esperan un razonamiento claro. Utilice normas armonizadas y definiciones coherentes siempre que sea posible. La Base de datos de terminología armonizada de CLSI ayuda a alinear la terminología en todos los documentos y comunicaciones.
Desafíos y errores comunes
Ambigüedad en el uso previsto: Las afirmaciones vagas desalinean los estudios y debilitan la evidencia.
Diseño de estudio subóptimo: Los estudios deben tener la potencia estadística adecuada con criterios de aceptación predefinidos.
Variables preanalíticas no controladas: Valide la recogida, el transporte y el almacenamiento; justifíquelos en el Plan de evaluación del rendimiento (PEP).
Deficiencias en la validación del software: Los DIV basados en algoritmos requieren controles del ciclo de vida del software (IEC 62304; IEC 82304-1).
Trazabilidad inadecuada: Vincule los datos brutos al Informe de rendimiento analítico (APR) y al Informe de evaluación del rendimiento (PER).
Supervisión del ciclo de vida: Los cambios en los reactivos, el software o los protocolos pueden desencadenar una revalidación. Mantenga el PMS y el PMPF para seguir cumpliendo la normativa.
Soluciones estratégicas
En MDx CRO, los equipos combinan experiencia regulatoria, científica y estadística para agilizar la validación analítica. Los servicios clave incluyen:
Evaluaciones de deficiencias con respecto a la guía IVDR, MDCG y CLSI.
Diseño de estudio analítico personalizado alineado con ISO 13485 y Anexo XIII.
Integración de PMS y PMPF en la gestión del ciclo de vida del rendimiento.
Conclusión
La validación analítica es más que una obligación regulatoria: forma la base de la credibilidad diagnóstica. Cuando se ejecuta bien, demuestra que un DIV es preciso, seguro y clínicamente eficaz. Con el IVDR aportando un mayor escrutinio, los fabricantes deben utilizar definiciones claras, justificaciones rigurosas y normas armonizadas para lograr una validación sólida y un éxito a largo plazo en el mercado.
¿Necesita ayuda para crear una validación analítica lista para auditorías según el IVDR? MDx CRO diseña estudios conformes, fortalece la trazabilidad y prepara archivos técnicos para revisiones más rápidas y fluidas. Póngase en contacto con nosotros hoy mismo.
Regulación de las pruebas de laboratorio desarrolladas (LDT) por la FDA
octubre 4, 2023
The FDA Laboratory Developed Tests regulation marks one of the most significant shifts in U.S. diagnostic oversight in decades. The FDA’s new rule phases in full regulation of LDTs over four years, with no grandfathering. This change elevates the importance of IVD CROs, whose regulatory and clinical expertise will be critical as laboratories adapt to stringent new requirements. The rule represents a major transformation in the U.S. IVD landscape and will reshape how laboratories develop, validate, and maintain LDTs.
Introduction
On September 29, 2023, the FDA released a groundbreaking proposed rule that fundamentally redefines how the agency regulates Laboratory‑Developed Tests (LDTs). This proposal shifts LDTs out of decades of enforcement discretion and brings them fully under the FDA’s medical device framework.
Because LDTs are a subset of in vitro diagnostic products (IVDs), the new rule has sweeping implications for clinical laboratories, manufacturers, and the broader diagnostics industry. Under the FDA Laboratory Developed Tests regulation, LDTs will now be treated like other medical devices—requiring quality systems, medical device reporting, registration, listing, and in many cases, premarket review.
For stakeholders across the IVD sector, this change is significant.
Key Points to Consider as the FDA regulates LDTs
Expanded Definition of IVDs The FDA proposes to explicitly classify LDTs as IVDs under 21 CFR 809.3. This means LDTs will now fall under the same requirements as traditional IVD medical devices.
Phased, Four‑Year Implementation The FDA will remove enforcement discretion in five stages over a four‑year timeline. Each stage introduces new regulatory obligations for laboratories.
No Grandfather Clause The proposal does not exempt existing LDTs. All LDTs (old and new) must eventually comply.
Test Categories Exempt from Enhanced Oversight Certain test types, including forensic tests and HLA assays, are proposed for exemption.
Public Comment Period Stakeholders were invited to submit comments through December 4, 2023.
Background on FDA Regulation for LDTs and IVDs
IVDs have traditionally been subject to rigorous regulatory scrutiny under various heads:
510(k) premarket notification or premarket approval (PMA)
Quality system regulation
Medical device reporting
Registration and listing
Labeling
LDTs, however, historically operated under enforcement discretion, receiving minimal oversight. This approach was based on the assumption that LDTs were low risk and used primarily within single laboratories.
That landscape has changed.
The Evolving Landscape of LDTs
Over the last 50 years, LDTs have become increasingly complex, widely used, and technically sophisticated. This evolution has driven demand for stronger oversight in areas such as:
Clinical validity
Analytical performance
Manufacturing consistency
Patient safety
The new FDA Laboratory Developed Tests regulation directly responds to these gaps. By redefining LDTs and removing enforcement discretion, the FDA aims to strengthen public health protections.
The Road Ahead: Key Regulatory Impacts
The phased implementation timeline will introduce major compliance requirements:
Medical Device Reporting
The first enforcement area to take effect.
Quality Systems Regulation
Expected three years after publication of the final rule.
Premarket Review
Introduced 3.5 to four years after the final rule, starting with high‑risk LDTs and expanding to moderate-and-low risk tests.
Labs performing LDTs must begin planning now. Clinical and analytical validation, documentation systems, and regulatory processes will all require upgrades.
Alignment With Europe’s IVDR Rollout
The FDA’s new approach mirrors developments in Europe under the In Vitro Diagnostic Regulation (IVDR). The IVDR already applies strict rules to in‑house tests and LDTs, requiring:
Adherence to Article 5.5 requirements for in‑house devices
çUnder IVDR, an LDT cannot be used if an equivalent CE‑marked test exists. This forces laboratories to justify in‑house development and meet near‑manufacturer‑level standards.
Conclusion: An Industry in Transition
As experts in IVD quality, regulatory, and clinical operations, MDx CRO encourages laboratories and manufacturers to prepare now for the FDA Laboratory Developed Tests regulation. Although legal challenges may influence the timeline, increased oversight is inevitable, and already fully established within Europe under the IVDR.
Stakeholders should submit comments to the FDA by December 4, 2023, and begin strengthening their regulatory systems immediately.
MDx: su CRO especializado en estudios clínicos de IVD en la UE
agosto 24, 2023
Introducción
En el mundo en rápida evolución del diagnóstico in vitro (DIV), los fabricantes comprenden cada vez más la necesidad de realizar estudios rigurosos del rendimiento clínico. Dichos estudios constituyen la columna vertebral para garantizar la seguridad, la eficiencia y la preparación general para el mercado de los dispositivos DIV. Con el estricto entorno regulatorio de la Unión Europea (UE), la realización de estos estudios requiere experiencia y precisión. Ahí es donde destaca MDx CRO, un nombre de confianza en la investigación por contrato de DIV y la consultoría regulatoria.
Toma de decisiones basada en evidencia: los estudios del rendimiento clínico proporcionan los datos que pueden probar la precisión diagnóstica, la precisión y la utilidad de los dispositivos DIV. Ayudan a los fabricantes a perfeccionar sus ofertas y justificar las afirmaciones de sus productos.
Cumplimiento normativo: garantizar el cumplimiento del Reglamento de la UE sobre productos sanitarios para diagnóstico in vitro (IVDR) y normas como la ISO 20916 es innegociable. Los estudios clínicos a menudo forman la base para obtener estas credenciales y abrir el mercado europeo.
Superando los desafíos con MDx CRO
Ya sea una empresa emergente o un gigante de los DIV, los desafíos como la selección del centro, el diseño del estudio, el seguimiento eficaz y el cumplimiento normativo pueden ser desalentadores. Aquí es donde MDx CRO puede ser su guía:
Experiencia comprobada: con su legado en el ámbito de los DIV y antiguos expertos de organismos notificados a bordo, MDx CRO ofrece información incomparable sobre el diseño de estudios eficaz, lo que garantiza que los fabricantes obtengan información práctica en todo momento.
Red de centros clínicos: gracias a sus años en la industria, MDx CRO ha establecido sólidas afiliaciones con centros clínicos líderes, lo que garantiza una realización del estudio oportuna y eficiente.
Conocimiento normativo: navegar por el laberinto de IVDR e ISO 20916 se vuelve más sencillo con el ala de consultoría regulatoria de MDx CRO, que garantiza que los fabricantes siempre cumplan con la ley.
Seguimiento integral: con un gran enfoque en los detalles, MDx CRO garantiza que cada estudio se mantenga encaminado, que se mantengan los protocolos y que la integridad de los datos permanezca intacta.
¿Por qué MDx CRO?
En pocas palabras, MDx CRO no es solo un proveedor de servicios, sino un socio en su viaje de DIV. Nuestro experimentado equipo comprende los desafíos únicos que enfrentan los fabricantes de DIV, lo que los convierte en un activo indispensable en el viaje de su producto desde el concepto hasta el mercado europeo.
Conclusión
Los estudios clínicos de DIV, aunque desafiantes, presentan una oportunidad de oro para que los fabricantes de DIV validen rigurosamente las afirmaciones de sus productos. En la intrincada red de regulaciones de la UE, los fabricantes necesitan más que solo experiencia; necesitan un socio. ¿Y quién mejor que MDx CRO, que ha demostrado constantemente excelencia en el diseño, el seguimiento y la garantía del cumplimiento normativo completo? Elija MDx CRO y trabajemos juntos para llevar dispositivos DIV transformadores y confiables al mercado de la UE.
Preguntas frecuentes sobre los estudios clínicos de DIV y MDx CRO:
¿Qué son los estudios clínicos de DIV?
Los estudios clínicos de DIV se refieren a investigaciones y evaluaciones rigurosas realizadas para determinar la seguridad, la eficiencia y el rendimiento general de los dispositivos de diagnóstico in vitro (DIV).
¿Por qué son importantes los estudios clínicos de DIV en la UE?
La UE tiene requisitos regulatorios estrictos. Los estudios clínicos de DIV proporcionan la evidencia necesaria para respaldar las afirmaciones de los productos, lo que garantiza el cumplimiento del Reglamento de la UE sobre productos sanitarios para diagnóstico in vitro (IVDR) y las normas internacionales como la ISO 20916.
¿Qué desafíos pueden esperar los fabricantes al realizar estudios de DIV en la UE?
Los fabricantes pueden enfrentar desafíos como la selección del centro, la creación de un diseño de estudio eficaz, el seguimiento regular del estudio y la garantía del cumplimiento de las regulaciones y normas de la UE.
¿Cómo ayuda MDx CRO con estos desafíos?
MDx CRO ofrece experiencia en el diseño de estudios, tiene afiliaciones con los principales centros clínicos, proporciona consultoría regulatoria para los estándares de la UE y garantiza el seguimiento integral del estudio para mantener la calidad y la integridad de los datos.
¿Es MDx CRO adecuado tanto para empresas emergentes como para fabricantes establecidos?
¡Absolutamente! Ya sea una empresa emergente que ingresa al mercado de DIV o un fabricante experimentado, las soluciones personalizadas de MDx CRO se adaptan a las necesidades únicas de cada cliente.
¿Cómo garantiza MDx CRO el cumplimiento de IVDR e ISO 20916?
MDx CRO cuenta con un ala de consultoría regulatoria con un profundo conocimiento de IVDR e ISO 20916, lo que garantiza que los fabricantes reciban orientación y asistencia precisas durante todo el viaje del dispositivo DIV al mercado. Nuestro equipo de antiguos expertos de organismos notificados a bordo ayuda a diseñar estudios que cumplan con las expectativas de la marca CE
¿Qué ventajas ofrece MDx CRO en términos de selección de centros para estudios de DIV?
Con su amplia experiencia y conexiones en la industria, MDx CRO ha establecido relaciones con centros clínicos líderes para una variedad de tecnologías y aplicaciones clínicas, lo que garantiza el inicio y la ejecución oportunos y eficientes del estudio.
¿Cómo afecta la asociación con MDx CRO a la tasa de éxito de los dispositivos DIV en el mercado de la UE?
Con los servicios integrales de MDx CRO, desde el diseño hasta el seguimiento y la orientación regulatoria, los fabricantes mejoran sus posibilidades de un lanzamiento de productos DIV exitoso y conforme en la UE.
¿Dónde puedo obtener más información sobre las historias de éxito o los estudios de caso de MDx CRO?
Lo mejor es comunicarse directamente con MDx CRO o visitar nuestro sitio web para obtener testimonios detallados, estudios de caso y más información sobre nuestro trabajo.
Written by:
Carlos Galamba
CEO
Líder sénior en materia de regulación y antiguo revisor de BSI IVDR con amplia experiencia en el marcado CE de IVD de alto riesgo, diagnósticos complementarios y la implementación de IVDR.
Consultoría de DIV in vitro para diagnóstico complementario dentro del marco de la EMA: guía exhaustiva
agosto 8, 2023
El campo de los diagnósticos complementarios DIV (CDx) representa una confluencia de innovación tecnológica, cumplimiento normativo y atención al paciente. A medida que la medicina personalizada se convierte en una parte integral de la atención médica, el marco regulatorio que rige los CDx, incluido el Reglamento sobre productos sanitarios para diagnóstico in vitro (IVDR), se ha vuelto más complejo. Este escenario exige una consultoría especializada en diagnósticos complementarios. MDx CRO está a la vanguardia de este ámbito, ofreciendo experiencia y orientación en el proceso de consulta de CDx con la Agencia Europea de Medicamentos (EMA), la preparación del organismo notificado y el cumplimiento del IVDR dentro de la Unión Europea (UE).
DIV de diagnósticos complementarios y su función
Los CDx son pruebas de diagnóstico in vitro (DIV) diseñadas para proporcionar información esencial para el uso seguro y eficaz de un medicamento correspondiente. Sus aplicaciones podrían incluir:
Identificar a los pacientes con más probabilidades de beneficiarse de un producto terapéutico en particular.
Determinar la idoneidad de los pacientes para tratamientos específicos.
Supervisar las respuestas a los tratamientos en curso.
El impacto del IVDR en los diagnósticos complementarios
El IVDR establece requisitos legales sólidos para los productos sanitarios para diagnóstico in vitro, incluidos los CDx. Los aspectos clave incluyen:
Mayor seguridad del paciente: Garantizar la calidad y la fiabilidad de los DIV de CDx.
Supervisión más estricta: Mayor escrutinio del proceso de desarrollo y aprobación de CDx. A diferencia de la directiva anterior, los CDx ahora requieren una evaluación de la conformidad por parte de un organismo notificado, una organización independiente designada para evaluar la conformidad de los productos sanitarios y los diagnósticos in vitro. Además, los CDx también son evaluados por una autoridad de medicamentos, muy probablemente la EMA (Agencia Europea de Medicamentos), pero también podría participar una autoridad competente.
Documentación técnica exhaustiva: Los mayores requisitos de evidencia clínica son particularmente notables en el IVDR. MDx CRO puede ayudar a los fabricantes de CDx y a sus socios farmacéuticos a recopilar los datos necesarios para respaldar su solicitud de CDx. Estos datos pueden incluir datos de ensayos clínicos (datos de rendimiento clínico), datos analíticos y datos de seguridad. Los fabricantes deben proporcionar evidencia clínica sólida para demostrar el rendimiento, la seguridad y la utilidad clínica del CDx.
Hay una serie de otros factores que pueden afectar el proceso de aprobación de CDx en la UE. Estos factores incluyen:
La disponibilidad de datos: Tanto el organismo notificado como la EMA deberán tener acceso a los datos de los ensayos clínicos que demuestren la seguridad y la eficacia del CDx.
La complejidad del CDx: Cuanto más complejo sea el CDx, más difícil será evaluar su seguridad y eficacia.
La novedad del CDx: Si el CDx implica nuevas tecnologías o indicaciones, la EMA y el organismo notificado deberán adoptar un enfoque más cauteloso para su aprobación. Diferentes escenarios influirán en el alcance del escrutinio involucrado, incluidos los escenarios de CDx codesarrollados, los CDx de seguimiento y los CDx que ya están en el mercado bajo la antigua directiva IVD.
Comprensión del procedimiento de consulta de diagnósticos complementarios de la EMA
El procedimiento de consulta lo inicia el organismo notificado cuando recibe una solicitud de un fabricante de CDx. El medicamento involucrado podría ser un medicamento ya autorizado para su comercialización en la UE o un medicamento en proceso de aprobación. La alineación de los procesos de desarrollo de fármacos y diagnósticos puede ayudar a garantizar que los resultados de los ensayos clínicos sean precisos y fiables, y que el medicamento sea seguro y eficaz cuando se utilice con el CDx.
La alineación de los plazos en el proceso de desarrollo de fármacos y diagnósticos (CDx) puede ayudar a garantizar que los ensayos clínicos del medicamento se lleven a cabo de una manera que sea coherente con el uso previsto del CDx.
Tras la solicitud de aprobación de un DIV de CDx, el organismo notificado presentará una carta de intención a la EMA, junto con un expediente técnico que describa el CDx y el medicamento.
A continuación, la EMA nombrará a un ponente, que será responsable de revisar el expediente técnico y emitir un dictamen científico sobre la idoneidad del CDx para su uso con el medicamento. El ponente también tendrá en cuenta las opiniones de otras partes interesadas, como el solicitante del medicamento, el fabricante del CDx y los grupos de pacientes.
La EMA proporcionará su dictamen científico sobre los aspectos del CDx que se relacionan con el medicamento al organismo notificado. A continuación, el organismo notificado utilizará el dictamen de la EMA para tomar una decisión sobre si concede o no el marcado CE al CDx, de acuerdo con los requisitos reglamentarios del reglamento sobre productos sanitarios para diagnóstico in vitro (EU IVDR).
Los plazos del procedimiento de la EMA desempeñan un papel importante en el éxito de la consulta y los plazos de respuesta pueden ser extremadamente cortos. Los fabricantes deben tener esto en cuenta al planificar sus presentaciones de CDx. Existe la posibilidad de solicitar una reunión previa a la presentación que incluirá a representantes de los organismos notificados, la EMA y también podría incluir al fabricante del fármaco; esto se utiliza estrictamente para alinear las consideraciones de procedimiento y plazos (no se utiliza para proporcionar comentarios sobre el diseño del estudio o el contenido de la documentación técnica).
Uno de los documentos clave utilizados en la consulta y presentado por el organismo notificado a la EMA es el SSP (Resumen de seguridad y rendimiento). La EMA espera que los fabricantes utilicen la plantilla SSP proporcionada en MDCG 2022-9. Se espera mucho más detalle en el SSP en comparación con la información proporcionada en el IFU. Por ejemplo, se necesitan detalles sobre los estudios de concordancia, particularmente para los CDx codesarrollados cuando se han utilizado diferentes versiones de un diagnóstico a lo largo del programa de desarrollo clínico.
MDx CRO: su socio en consultoría de diagnósticos complementarios
Nuestros servicios de consultoría de diagnósticos complementarios abarcan todas las etapas de desarrollo, aprobación y vigilancia posterior a la comercialización:
Orientación sobre los requisitos del IVDR: Apoyo en profundidad para comprender y cumplir las demandas específicas del IVDR en lo que respecta a los CDx. MDx CRO puede ayudar a una empresa de diagnóstico a identificar los requisitos específicos que se aplican a su CDx. Por ejemplo, los requisitos para un CDx que está destinado a evaluar la idoneidad de un paciente para el tratamiento pueden ser diferentes de los requisitos para un CDx que está destinado a utilizarse para supervisar la respuesta de un paciente al tratamiento.
Preparación para la evaluación del organismo notificado: Estrategias personalizadas para la evaluación exitosa de un CDx bajo el IVDR: Asistencia para recopilar y presentar la documentación técnica necesaria y los documentos relacionados con la calidad.
Proporcionar formación al personal del fabricante: MDx CRO puede proporcionar formación al personal del fabricante sobre los requisitos de la EMA para los CDx, así como sobre el proceso de evaluación y las expectativas del organismo notificado. Esta formación ayudará a garantizar que el personal del fabricante esté preparado para responder a cualquier pregunta del organismo notificado y aumentará las posibilidades de éxito.
Comunicación con las partes interesadas: Facilitar la comunicación con todas las partes relevantes.
Perspectiva global: Navegar por las consideraciones internacionales para los CDx en estudios de varios países.
Soporte posterior a la comercialización: Centrado en mantener los más altos estándares a través de la supervisión continua del cumplimiento del IVDR y otros requisitos reglamentarios. Esto incluye la implementación de procesos sólidos de vigilancia posterior a la comercialización y evaluaciones de seguimiento del rendimiento posterior a la comercialización (PMPF), la supervisión del rendimiento del CDx en entornos clínicos del mundo real, el seguimiento y el análisis de los eventos adversos relacionados con el uso del CDx y la realización de estudios continuos para evaluar el impacto y la eficacia a largo plazo del CDx.
¿Por qué MDx CRO para la consultoría de DIV de diagnósticos complementarios?
Experiencia: Nuestro profundo conocimiento de los CDx, el IVDR y las regulaciones de la UE ofrece un apoyo sin igual.
Colaboración: Trabajando en estrecha colaboración con los clientes, adaptamos nuestro enfoque para satisfacer las necesidades específicas.
Eficiencia: Nuestros conocimientos y orientación ahorran tiempo y recursos valiosos, simplificando las complejas vías regulatorias.
Compromiso: Nuestra dedicación a la excelencia, la seguridad del paciente y la innovación nos distingue.
Navegar por el mundo multifacético de los diagnósticos complementarios en la UE, con la complejidad añadida del IVDR, requiere un socio dedicado y cualificado. MDx CRO está listo para ser su guía en este viaje crítico, asegurando la alineación con todos los estándares regulatorios. Póngase en contacto para explorar cómo nuestra consultoría de diagnósticos complementarios puede ser la clave para desbloquear su potencial de CDx en el entorno regulatorio dinámico de la UE.
Preguntas frecuentes
P: ¿Qué es un diagnóstico complementario codesarrollado en el contexto de la consulta de la EMA?
R: Un CDx codesarrollado es un dispositivo desarrollado junto con un medicamento para la autorización inicial o un cambio de indicación. Esto puede incluir el desarrollo durante un ensayo clínico fundamental o un estudio puente, con documentación suficiente para garantizar la alineación del rendimiento.
P: ¿En qué se diferencia un CDx de seguimiento de un CDx codesarrollado?
R: Un CDx de seguimiento busca la misma indicación que el CDx original, pero no se desarrolla en paralelo con el medicamento. El CDx de seguimiento se dirige al mismo biomarcador, pero puede no basarse en la misma tecnología. Debe ser muy comparable al original en rendimiento, seguridad y eficacia.
P: ¿Qué documentación se requiere para un CDx de seguimiento?
R: Se debe proporcionar documentación suficiente para un CDx de seguimiento para demostrar que su rendimiento analítico es comparable al CDx original y que no hay ningún impacto en el rendimiento clínico incompatible con el uso seguro y eficaz del medicamento.
P: ¿Cómo se gestionan los dispositivos que hacen la transición de IVDD a IVDR?
R: Los dispositivos comercializados inicialmente bajo la Directiva 98/79/CE (IVDD) que hacen la transición a IVDR se incluyen en los escenarios codesarrollados o de seguimiento, dependiendo de cómo se desarrollaron inicialmente.
P: ¿Es posible proceder con un único procedimiento de consulta de CDx para múltiples medicamentos e indicaciones autorizados?
R: Sí, si el propósito previsto de un dispositivo incluye varios medicamentos e indicaciones autorizados, se recomienda proceder con un único procedimiento de consulta de CDx. Todos los medicamentos afectados deben figurar en la carta de intención de presentación del organismo notificado y en el formulario de solicitud.
Written by:
Carlos Galamba
CEO
Líder sénior en materia de regulación y antiguo revisor de BSI IVDR con amplia experiencia en el marcado CE de IVD de alto riesgo, diagnósticos complementarios y la implementación de IVDR.
Desarrollo de software IVD: cómo comercializar software IVD en 8 pasos
abril 4, 2023
El sector sanitario está experimentando una rápida transformación impulsada por la llegada de tecnología de diagnóstico médico avanzada. El desarrollo de software IVD es un componente fundamental de esta revolución, ya que permite realizar pruebas, análisis, informes y comunicación sin necesidad de un laboratorio físico ni de una visita al consultorio médico.
Llevar el desarrollo de software IVD al mercado puede beneficiar a los pacientes y a los proveedores de atención médica, que pueden brindar atención de calidad más rápido y con menos recursos.
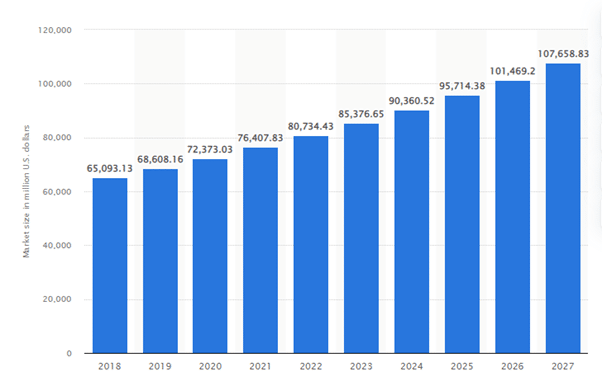
Tamaño proyectado del mercado de IVD en todo el mundo de 2018 a 2027 (en millones de dólares estadounidenses)
El gráfico anterior muestra que el mercado de diagnóstico in vitro (IVD) a nivel mundial se estimó en 72 400 millones de dólares estadounidenses en 2020, con un crecimiento proyectado de 108 000 millones de dólares estadounidenses para 2027, lo que demuestra su mayor relevancia en el sector sanitario actual.
1. Realizar estudios de mercado
Antes de iniciar el proceso de desarrollo, las organizaciones deben realizar estudios de mercado y comprender su mercado objetivo, las necesidades de los usuarios y los posibles competidores.
La investigación de mercado también debería ayudar a determinar los requisitos reglamentarios que las organizaciones deben cumplir y las tendencias actuales dentro del sector o el espacio tecnológico.
A continuación, se indican algunos puntos clave que debe tener en cuenta al realizar estudios de mercado para el desarrollo de software IVD:
Identificar los requisitos reglamentarios: Identificar los requisitos reglamentarios de su software es esencial para comercializar su producto. Pueden variar en función de su mercado objetivo, como la FDA en los EE. UU. o los requisitos de marcado CE de la Unión Europea (IVDR 746/2017).
Determinar su mercado objetivo: Identifique los segmentos del sector sanitario a los que servirá su software IVD. Tenga en cuenta factores como la geografía, el tipo de organización sanitaria y las áreas de especialidad.
Identificar a sus competidores: Investigue el mercado de software IVD para identificar a sus competidores y sus productos. Analice sus puntos fuertes, débiles, precios y estrategias de marketing.
Comprender a sus clientes: Realice encuestas, entrevistas y grupos focales con profesionales sanitarios para comprender sus necesidades, preferencias y puntos débiles. Utilice esta información para adaptar su software IVD a sus necesidades específicas.
Analizar las tendencias del mercado: Manténgase al día de las últimas tendencias y novedades en el mercado de software IVD. Supervise las publicaciones del sector, asista a conferencias y siga a expertos del sector y líderes de opinión en las redes sociales.
Determinar la estrategia de precios: Tenga en cuenta los costes, el mercado objetivo y la competencia al determinar la estrategia de precios, por ejemplo, una tarifa única. ¿Cuota de suscripción? ¿Tarifa por prueba?
2. Clasificación del software IVD
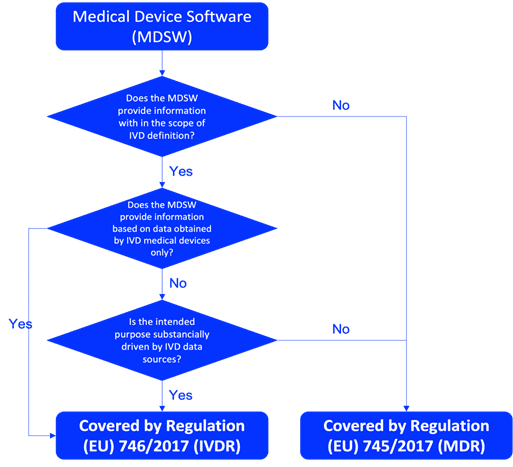
Comprender la clasificación de su software IVD es fundamental antes de iniciar el proceso de planificación y diseño. Determine si el software IVD es un dispositivo médico IVD independiente o un componente de un sistema más grande. Para ser calificado como software de dispositivo médico IVD en la UE, el producto debe cumplir con la definición de IVD de acuerdo con el Artículo 2(2) del Reglamento IVDR746/2017, como se describe en la Figura 1. La clasificación correcta del software IVD debe realizarse en función de las reglas descritas en el Anexo VIII del IVDR. Los documentos de orientación pertinentes, como MDCG 2019-11, también son esenciales para la calificación y clasificación del software IVD.
Fig. 1: Diagrama de flujo de MDCG 2019-11 sobre la calificación del software de dispositivos médicos (MDSW)
Familiarícese con los marcos regulatorios, las guías y los estándares relevantes, como ISO 13485, IEC 62304, pero también con los documentos de guía específicos publicados por los reguladores, que brindan especificaciones y pautas para desarrollar, validar y mantener el software IVD.
3. Planificar y diseñar el software
El siguiente paso crucial para un desarrollo de software IVD exitoso es el diseño y la planificación.
Un proceso de planificación sólido y bien documentado puede ayudar a proporcionar una hoja de ruta más detallada para el desarrollo.
Durante esta fase, las revisiones de diseño, las pruebas y la verificación garantizarán que la versión final cumpla con los requisitos del usuario.
Es esencial incorporar los comentarios de los usuarios en cada etapa del proceso de diseño para desarrollar una interfaz intuitiva que funcione eficazmente de acuerdo con sus necesidades.
Obtener comentarios de los clientes y las partes interesadas ofrece al equipo de desarrollo oportunidades para reconocer posibles inquietudes y áreas de mejora.
Los desarrolladores pueden idear soluciones de software que satisfagan las necesidades de los clientes y aborden sus quejas integrando los comentarios.
No se debe subestimar la importancia de una documentación precisa, ya que ayuda a rastrear los problemas más adelante en el ciclo de vida del software.
El equipo de desarrollo debe tener en cuenta la escalabilidad y la flexibilidad durante las etapas iniciales de planificación y diseño al crear el software.
4. Desarrollar y probar el software
Desarrollar y probar el software es crucial para crear un prototipo funcional.
La fase de desarrollo es necesaria para garantizar la precisión del diseño, la codificación y los algoritmos utilizados en la creación de las soluciones de software.
Las pruebas y el control de calidad también desempeñan un papel esencial para garantizar que los productos cumplan con todos los requisitos antes del lanzamiento.
Es esencial que las empresas evalúen minuciosamente cada componente de su software como parte de este proceso. Esto incluye garantizar que cumplan con los objetivos de rendimiento con respecto a la velocidad, la capacidad de respuesta, la escalabilidad, la seguridad, la fiabilidad y la facilidad de uso para sus usuarios.
Los controles de garantía de calidad ayudan a identificar errores o fallos para lanzar un producto sin defectos que cumpla con todos los estándares de organismos reguladores como la FDA o el marcado CE.
Al implementar sistemas IVD, los fabricantes deben considerar si sus aplicaciones pueden ser lo suficientemente flexibles para admitir nuevos avances tecnológicos; la preparación para el futuro de sus productos se vuelve cada vez más necesaria donde los clientes exigen longevidad en las actualizaciones o iteraciones a lo largo del tiempo.
5. Preparar una presentación reglamentaria
Preparar un paquete de presentación reglamentaria es fundamental para comercializar el desarrollo de software IVD. Este paso implica la recopilación de documentos para demostrar que el software cumple con los requisitos reglamentarios y es seguro y eficaz.
Estas son algunas consideraciones críticas para preparar una presentación reglamentaria:
Recopilar los documentos pertinentes para el paquete
Comprender las regulaciones, los estándares y la clasificación de riesgo del software IVD y el rol de fabricación. Los documentos clave que se deben incluir son la descripción del dispositivo, la documentación técnica, el archivo de gestión de riesgos (ISO 14971), la documentación del ciclo de vida del software (IEC 62304) y la documentación del sistema de gestión de calidad (ISO 13485). La gestión de riesgos debe aplicarse y supervisarse durante el ciclo de vida del desarrollo de software IVD.
Preparar un informe de evaluación del rendimiento (PER)
Esto requerirá un análisis exhaustivo de la evidencia científica que demuestre que el producto satisface las necesidades del usuario de forma segura y eficaz. La evaluación del rendimiento del software IVD debe prepararse de acuerdo con los documentos de orientación pertinentes, como MDCG 2020-1 para la UE. Otra guía, como MedTech Europe Requisitos de evidencia clínica para IVD, también puede ser una buena fuente de información adicional.
Los estudios de rendimiento clínico tienen como objetivo proporcionar evidencia de la seguridad y eficacia del propósito previsto de un producto para garantizar que pueda diagnosticar, controlar y predecir enfermedades y afecciones con precisión.
Como se describe en MDCG 2020-1“La validación del rendimiento clínico debe considerarse en cada cambio del software a una nueva versión. Si no se realiza ninguna validación, se debe indicar una justificación en la documentación técnica. Con una validación del rendimiento clínico, se demuestra que los usuarios pueden lograr resultados clínicamente relevantes a través del uso predecible y fiable del MDSW”.
La adhesión a las normas y directrices pertinentes, como ISO 20916 (Estudios de rendimiento clínico para diagnósticos in vitro) y las buenas prácticas clínicas (CGP), es crucial para la ejecución exitosa de los estudios clínicos.
Garantizar la precisión de los datos
Asegúrese de que todos los datos recopilados de las pruebas se presenten con precisión para demostrar la seguridad y la eficacia antes de enviar su solicitud. Esto incluye los datos de validación y verificación, las evaluaciones de rendimiento y, si corresponde, los resultados de los estudios clínicos. Revise cuidadosamente toda la información para verificar su exactitud e integridad antes de enviarla.
6. Obtener la aprobación reglamentaria
Debe obtener la aprobación reglamentaria para comercializar el desarrollo de software IVD. Una aprobación exitosa le permite comercializar y vender su software de conformidad con las leyes y regulaciones locales.
Familiarícese con los marcos y directrices regulatorias aplicables para su software. Estos pueden incluir ISO 13485 (sistemas de gestión de calidad), IEC 62304 (procesos del ciclo de vida del software de dispositivos médicos), ISO 14971 (gestión de riesgos) y documentos de orientación de MDCG como MDCG 2019-11 (calificación y clasificación del software en las regulaciones de dispositivos médicos) y MDCG 2020-1 (guía sobre la evaluación del rendimiento para el software IVD según el IVDR. También debe considerar las directrices de otras jurisdicciones según su estrategia de mercado. La FDA, por ejemplo, ha emitido guías para el software como dispositivo médico (SAMD).
Desarrolle un paquete de solicitud integral que incluya todos los documentos, datos y pruebas necesarios para la revisión. Asegúrese de que su paquete aborde los requisitos reglamentarios, como los procedimientos de evaluación de la conformidad, la evidencia clínica y la vigilancia posterior a la comercialización.
Envíe el paquete de solicitud a las autoridades reguladoras pertinentes para su revisión, como la FDA en los EE. UU. o los organismos notificados en la UE en virtud del Reglamento de diagnóstico in vitro (IVDR) 2017/746.
Esté preparado para responder de forma rápida y precisa a cualquier comentario o solicitud de información adicional de las agencias reguladoras durante el proceso de revisión.
Realice los cambios recomendados rápidamente como parte de su presentación para obtener la aprobación de las agencias con éxito.
Una vez que reciba la aprobación de las agencias reguladoras, puede seguir adelante con la comercialización y venta de su software de acuerdo con las leyes y regulaciones locales, asegurando el cumplimiento continuo de cualquier requisito posterior a la comercialización.
7. Desarrollo de estrategias de marketing y ventas
Crear una estrategia de marketing y ventas exitosa es esencial para llevar el software IVD al mercado, ya que permite un posicionamiento más rápido y obtener una ventaja competitiva. Asegúrese de desarrollar una identidad de marca sólida con mensajes que resuenen con su audiencia.
Además, investigar las necesidades de los clientes y comprender las tendencias clave de la industria puede crear un enfoque más específico cuando se trata de la comercialización de soluciones de software IVD, lo que aumenta su probabilidad de éxito.
Asegúrese de utilizar múltiples canales, como publicidad de pago, campañas de correo electrónico, redes sociales y seminarios web, para llegar a clientes potenciales de diversos segmentos.
Y, por último, pero no menos importante, la creación de estrategias de comunicación eficaces para interactuar con los clientes durante todo el ciclo de ventas también será clave para promocionar los productos IVD con éxito.
8. Lanzar y dar soporte al software
Lanzar y dar soporte al software es un elemento crucial para su éxito. El producto se puede mejorar con el tiempo proporcionando actualizaciones periódicas y servicio al cliente, y los usuarios pueden obtener la mejor experiencia.
Estos son algunos puntos que debe tener en cuenta al lanzar su desarrollo de software IVD:
Cree un plan de soporte integral que ponga las necesidades del cliente en primer lugar. Asegúrese de tener un proceso eficiente para gestionar las consultas y los problemas técnicos a medida que surjan.
Asegúrese de que todas las actualizaciones de software necesarias se completen según lo programado, para que los usuarios no experimenten ningún retraso en el acceso a las funciones completas del producto o las correcciones de errores.
Evalúe el impacto regulatorio de los cambios y las correcciones de errores, los cambios en su software IVD pueden ser o no significativos. Considere si los cambios impactan sus aprobaciones regulatorias. MDCG 2022-6 proporciona orientación adicional sobre los cambios en el diseño y el propósito previsto en el contexto de los nuevos plazos de transición para los IVD en Europa.
Configure formularios de comentarios de los usuarios o encuestas para que los clientes puedan compartir sus opiniones sobre el rendimiento del producto y las mejoras que desean ver. Esto ayudará a impulsar un mayor desarrollo del software con el tiempo.
Ofrezca oportunidades de formación continua para las nuevas funciones, para que los usuarios se sientan seguros al utilizarlas una vez que se publiquen. Esto también garantizará que los clientes sepan cómo utilizar plenamente su inversión en su solución de desarrollo de software IVD.
Revolucionando la atención médica con el desarrollo de software IVD
El desarrollo de software de diagnóstico in vitro (IVD) ha transformado el sector sanitario al proporcionar soluciones rentables de pruebas, análisis, informes y comunicación sin equipos de laboratorio físicos.
Los estudios indican que los proveedores de atención médica dan alta prioridad a los procedimientos de diagnóstico in vitro (IVD), y su optimización tiene el potencial de mejorar los resultados de los pacientes. Por lo tanto, el desarrollo de software IVD es crucial para facilitar resultados de diagnóstico más rápidos y precisos, lo que en última instancia conduce a la optimización de las prácticas de atención médica.
Si necesita un socio en el desarrollo de software IVD para su negocio, MDx CRO es una consultoría de IVD que proporciona soluciones integrales para acompañarle en cada paso del proceso. Nuestro equipo de estrategas de CRO altamente experimentados tiene una amplia experiencia en llevar dispositivos médicos innovadores y tecnologías IVD al mercado. Solicite hoy mismo su consulta experta.
Preguntas frecuentes
¿Cuáles son las consideraciones clave al diseñar software IVD?
Hay varias consideraciones clave que las empresas deben tener en cuenta al diseñar software IVD: los requisitos del usuario, los requisitos reglamentarios según la ubicación geográfica de destino, la precisión de los datos y la gestión eficaz de los datos, la capacidad del software para integrarse con otros sistemas, así como el rendimiento y la usabilidad.
¿Cuáles son los requisitos reglamentarios para el desarrollo de software IVD en Europa?
Los requisitos reglamentarios para el desarrollo de software IVD en Europa están determinados por el Reglamento de diagnóstico in vitro (IVDR), que se hizo aplicable el 26 de mayo de 2022. Incluyen, entre otros, el diseño y el desarrollo, la gestión de riesgos, la validación y la verificación, así como el cumplimiento del RGPD.
¿Cuáles son los desafíos más comunes en el desarrollo de software IVD?
Los desafíos más comunes en el desarrollo de software IVD incluyen el cumplimiento normativo (que puede ser complejo y difícil de superar), garantizar la compatibilidad de la integración con otros sistemas, la gestión eficaz de los datos y una gran experiencia de usuario, entre otros.
¿Cómo se garantiza la calidad y la fiabilidad del software IVD?
Para garantizar la calidad y la fiabilidad del software IVD, es importante que las empresas sigan todas las directrices reglamentarias aplicables a su ubicación geográfica y utilicen un sistema de gestión de calidad para garantizar que el proceso de desarrollo esté bien documentado. La realización de pruebas, la validación y los procesos de verificación es otro elemento esencial del desarrollo de software para el diagnóstico in vitro.
¿Qué se debe tener en cuenta al desarrollar un estudio de rendimiento clínico de DIV para el cumplimiento del IVDR?
marzo 9, 2023
Los dispositivos de diagnóstico in vitro (DIV) son esenciales en la atención médica, ya que brindan información de diagnóstico precisa y confiable a los proveedores de atención médica. El desarrollo de un dispositivo DIV implica varias etapas, que incluyen investigación y desarrollo, diseño y creación de prototipos, verificación y validación, aprobación regulatoria y comercialización.
Uno de los pasos críticos en el desarrollo de DIV es la realización de un estudio de rendimiento clínico IVDR para generar datos fiables y significativos que respalden la aprobación regulatoria y el éxito comercial del dispositivo. En Europa, el reglamento de diagnóstico in vitro (EU IVDR) ya está en vigor y todos los productos nuevos que se comercialicen deben cumplir con requisitos muy estrictos de rendimiento clínico.
El papel de la norma ISO 20916 en los estudios de rendimiento clínico IVDR
El diseño y la ejecución de un estudio de rendimiento clínico de DIV son fundamentales para su éxito, y se deben considerar varios factores para garantizar que el estudio genere datos fiables y significativos. La Organización Internacional de Normalización (ISO) ha desarrollado la ISO 20916, una norma que proporciona orientación sobre el diseño y la realización de estudios clínicos para dispositivos médicos DIV. El objetivo de la norma es ayudar a los fabricantes, reguladores y otras partes interesadas a garantizar que los estudios de rendimiento clínico de DIV se diseñen y realicen de manera coherente y científicamente rigurosa.
La norma ISO 20916 abarca varios aspectos importantes, como el diseño del estudio, la determinación del tamaño de la muestra, la selección de criterios de valoración adecuados, el análisis estadístico y la presentación de informes de los resultados del estudio. La norma enfatiza la importancia de diseñar estudios que sean apropiados para el uso previsto del dispositivo DIV y que incorporen los principios de buenas prácticas clínicas (BPC).
El plan debe especificar los objetivos del estudio, los criterios de inclusión y exclusión para los participantes del estudio, los criterios de valoración del estudio y el plan de análisis estadístico, entre muchos otros requisitos. También debe incluir procedimientos para la gestión de datos y el control de calidad para garantizar la precisión y fiabilidad de los datos recopilados.
Otro aspecto importante de la norma ISO 20916 es el requisito de garantizar la seguridad y el bienestar de los participantes en el estudio. La norma enfatiza la importancia de obtener el consentimiento informado de los participantes en el estudio y de proteger su privacidad y confidencialidad. La norma también exige que los estudios se realicen de conformidad con los principios éticos y los requisitos reglamentarios.
Alineación con el EU IVDR
Además de la norma ISO 20916, la implementación del EU IVDR ha aumentado la importancia de realizar estudios de rendimiento clínico de DIV, ya que son necesarios para el cumplimiento normativo. El IVDR reemplazó a la anterior Directiva sobre productos sanitarios para diagnóstico in vitro (IVDD) e introdujo requisitos más estrictos para los dispositivos DIV, incluidos los requisitos de evidencia clínica. Ahora se exige a los fabricantes de DIV que demuestren evidencia clínica que respalde el propósito previsto de un dispositivo y su seguridad y rendimiento. Esto es particularmente importante, porque una evidencia clínica insuficiente podría conducir en última instancia a un rechazo del producto en el Organismo Notificado, lo que resultaría en costos adicionales y retrasos en la comercialización del producto.
Entre muchos requisitos, un estudio de rendimiento clínico IVDR se diseña y se lleva a cabo de conformidad con los principios de BPC. La norma ISO 20916 tiene requisitos adicionales, y tanto el reglamento como la norma deben ser considerados por todos los fabricantes de diagnósticos al desarrollar planes o protocolos de estudio de rendimiento clínico.
¿Cómo puede ayudar MDx CRO?
MDx es una organización de investigación por contrato (CRO) de dispositivos médicos y DIV que puede ayudar a los fabricantes de dispositivos DIV con sus estudios de rendimiento clínico proporcionando una gama de servicios, que incluyen:
diseño del estudio
selección del sitio
reclutamiento de pacientes
monitorización del estudio
gestión de datos
análisis estadístico
MDx tiene una amplia experiencia en la realización de estudios de rendimiento clínico para dispositivos DIV y un profundo conocimiento de los requisitos reglamentarios para estos estudios. Nuestro equipo de profesionales está bien capacitado y tiene experiencia en la gestión de todos los aspectos del estudio, desde el desarrollo del protocolo hasta la ejecución del estudio y el análisis de datos. Trabajamos en estrecha colaboración con nuestros clientes para garantizar que sus estudios se diseñen y se lleven a cabo de conformidad con las normas y directrices aplicables y que generen datos fiables y significativos.
Conclusión
La realización de un estudio de rendimiento clínico de DIV es un paso fundamental en el desarrollo y la comercialización de un dispositivo DIV. Siguiendo las mejores prácticas, trabajando con profesionales experimentados y seleccionando la CRO adecuada, los fabricantes de dispositivos DIV pueden generar datos fiables y significativos que pueden respaldar la aprobación regulatoria y el éxito comercial del dispositivo, lo que en última instancia beneficia a los pacientes y a los proveedores de atención médica. El cumplimiento del IVDR y de la norma ISO 20916 puede ayudar a garantizar que los datos generados sean aceptables para la presentación regulatoria y cumplan con los requisitos de seguridad y rendimiento para los DIV.
Solicite el paquete de servicios de medicina de precisión
Obtenga acceso al paquete de servicios de medicina de precisión de MDx, que detalla nuestras capacidades en investigación clínica, estrategia regulatoria y diagnósticos complementarios. Descubra cómo apoyamos a las empresas farmacéuticas, de CDx y a los desarrolladores de terapias avanzadas a lo largo del ciclo de vida clínico y regulatorio.